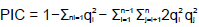ABSTRACT
Taro (Colocasia esculenta. (L.) Schott) is a genus of perennial plants that is widely distributed in the tropics or subtropics of Asia, Africa and America, which is the fourteenth most consumed vegetable of the world. However, molecular genetic research of Colocasia has been hindered by the insufficient genomic and transcriptome information. Here, the transcriptome of taro variety ‘Jingjiang Xiangsha’ from Jiangsu, China, was sequenced using the Illumina HiSeqTM 2000 platform in 2015. A total of 58,263,364 reads were generated, and assembly resolved into 65,878 unigenes with a N50 length of 1,357 bp. A total of 40,375 unigene sequences were successfully annotated based on searches against six public databases. Among the annotated unigenes, 14,753 were identified by gene Ontologyterms, 16,643 were classified to Clusters of Orthologous Groups categories, and 25,401 were mapped to 127 pathways in the Kyoto Encyclopedia of genes and genomes database. Also, 11,363 potential microsatellite loci were identified in 5,671 unigenes, and 150 primer pairs were randomly selected and amplified in 18 accessions of C. esculenta. A total of 100 primer pairs showed polymorphisms in repeat length. The number of alleles per locus ranged from 2 to 8. Across the 100 microsatellite loci, the polymorphism information content values ranged from 0.042 to 0.778. The transcriptomic data and microsatellite markers will play important roles in future functional gene analyses and genetic map construction of taro.
Key words: Colocasia, genetic diversity, microsatellite markers, transcriptome.
Taro (Colocasia esculenta L. Schott) is a major root crop belonging to the family Araceae. This plant originates from tropical Asia and America, and has been cultivated and utilized as food source and a medicinal herb in China for 2,000 years, which is also the fourteenth most consumed vegetable worldwide as a staple source of diets with high yield and nutritional value for people around the world (Ramanatha et al., 2010). Until now, researches on C. esculenta mainly focus on the biological characteristics, morphological variations, resistance and economic performance (Ahmed et al., 2013; Hunt et al., 2013; Nath et al., 2014; Das and Das, 2014; Das et al., 2015; Doungous et al., 2015; Soulard et al., 2016; Oliveira et al., 2017).
Microsatellites are a special class of repetitive DNA sequences that are distributed throughout the genome, and gradually became preferred markers for many applications in genetics and genomics (Chaïr et al., 2016; Dai et al., 2016; Vandenbroucke et al., 2016). However, the applications of microsatellite markers require reference genome and transcriptome data, thus, the development of microsatellite markers for non-model species, such as C. esculenta, are blocked by high cost and technical difficulties. RNA sequencing (RNA-Seq) is a high-throughput method to obtain large amounts of transcriptome sequence information, which is also effective for non-model organisms that lack a reference genome (Ellegren 2014; Waples et al., 2016). Transcriptome sequences include only encoding sequences, from which a high quality of functional information can help to reveal the molecular mechanisms and genetic maps (Fu et al., 2013; Hause et al., 2016).
In addition, transcriptome data is feasible for a large-scale development of microsatellite markers. Compared with genomic simple sequence repeat (SSR) markers, genetic markers developed based on RNA-Seq technologies provide a high efficiency method to identify candidate functional genes. Moreover, the continuous improvements in next-generation sequencing (NGS) technologies have made it a simple, economical, and reliable approach for novel gene discovery, molecular mechanisms analysis and molecular marker-assisted selection in many non-model organisms (He et al., 2014). Here, the Illumina HiSeq™ 2000 platform was used to characterize the transcriptome of C. esculenta. The transcriptome sequences were assembled and annotated based on searches against public databases. Microsatellites in the transcript sequences were detected in silico and SSR markers were randomly selected for validation experiment. The transcript sequence information and available SSR markers will provide valuable resources for further genetic and breeding research of C. esculenta.
Plant material and RNA extraction
For the RNA required for the transcriptome sequencing, leaves, stems and corms of taro variety ‘Jingjiang Xiangsha’ (Figure 1) were sampled from three 150-day-old plants, which were planted in the greenhouse under a 14/10 h photoperiod at 25°C (day) and 20°C (night) in Institute of Industrial Crops, Jiangsu Academy of Agricultural Sciences. Fresh leaves of 18 accessions of C. esculenta were collected for the validation of polymorphic SSR markers (Table 1). All samples were snap-frozen in liquid nitrogen and stored at −70°C. A Total RNA isolation system (Takara, Japan) was employed to extract RNA from tissues of the 150-day-old plant, following the manufacturer’s instructions. The quality of the RNA Integrity Number (RIN) was verified using a 2100 Bioanalyzer RNA Nanochip (Agilent, Santa Clara, CA) and its concentration ascertained using a ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE). The standards applied were 1.8≤ OD260/280≤2.2 and OD260/230≥2.0. At least 20 µg of RNA was pooled in an equal amount from each leaves, stems and corms used.

cDNA library construction and sequencing
Illumina (San Diego, CA) sequencing was performed at the Genomics Institute (Wuhan, China;
http://www.genomics.cn/index.php), following the manufacturer’s protocols. Poly (A) mRNA from the total RNA was isolated from generated fragments in the size range of 100–400 bp. The resulting fragments served as a template for the synthesis of the first strand cDNA. And then, second strand cDNA was synthesized and purified. The products were ligated through sequencing adapters, and sequenced using an Illumina HiSeq
TM 2000 device.
De novo assembly and gene annotation
Image data output from the sequencing device were transformed into raw reads and stored in FASTQ format. After filtered, the adapter and low quality sequences and the assembly of the transcriptome was achieved using the short-read assembly program Trinity. The unigenes are divided into either clusters or singletons. BLASTX alignments (applying an E-value of less than 10
−5) were performed between each unigene sequence and those lodged in non-redundant protein database (Nr, NCBI), non-redundant nucleotide database (Nt, NCBI), swiss-prot, gene ontology (GO,
http://www.geneontology.org/), clusters of orthologous groups (COG) databases (
http://www.ncbi.nlm.nih.gov/COG/), kyoto encyclopedia of genes and genomes (KEGG) and pathway database (
http://www.genome.jp/tools/kaas/). Functional annotation was assigned using the protein (Nr and Swiss-Prot), COG, GO and KEGG databases. BLASTX was employed to identify related sequences in the protein databases based on an E-value of less than 10
−5. The annotations acquired from Nr were processed through the Blast2GO program to obtain the relevant GO terms, and these were then analyzed by WEGO software to assign a GO functional classification and to illustrate the distribution of gene functions.
Microsatellite marker development and primer design
Microsatellite loci were identified by the simple sequence repeat identification software MISA (MIcroSAtellite identification tool) (
http://pgrc.ipk-gatersleben.de/misa/), applying the following parameters: a minimum of six repeats for dinucleotide motifs, of five for tri, of four for tetra, and of three for penta- and hexa-nucleotides. Appropriate primers of SSRs were designed through Primer 3.0 software (
http://sourceforge.net/projects/primer3), based on the following criteria: primer length 18 to 22 bp (optimally 20 bp), T
m of 50 to 60°C (no more than a 4°C difference between the T
ms of the forward and reverse primers) and an amplicon length in the range 100 to 400 bp. All primers were synthesized by Genscript (Nanjing, China).
SSR polymorphism validation and data analysis
Eighteen accessions of C. esculenta were selected for polymorphism validation of microsatellite loci. 100 primer pairs were randomly selected. Genomic DNA was isolated from leaves using the standard phenol–chloroform protocol (Hunt et al., 2013). PCR ampliï¬cation was performed on a gradient thermal cycler (Bio-Rad) with the following protocol: denaturation for 3 min at 95°C; 36 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 30 s; and finally 72°C for 7 min as an extension step. Finally, the PCR products were initially assessed for size polymorphisms on 6% denaturing polyacrylamide gels and then visualized by silver nitrate staining. The genetic information and indexing of polymorphic microsatellite loci were calculated using POPGENE 1.31 (Yeh et al., 2000) and PowerMarker v3.25 (Liu and Muse, 2005). Polymorphism information content (PIC) was derived using the following formula:
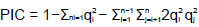
Where qi and qj represent the frequency of the ith and jth alleles, and n is the total number of alleles detected for a given SSR marker (Shete et al., 2000).
Sequencing and de novo assembly
RNA-Seq technology was used to sequence a pooled cDNA library of taro variety ‘Jingjiang Xiangsha’. The sequencing yielded approximately 58,263,364 raw pair-ended reads with a length of 100 bp. The raw read files were deposited in the NCBI Sequences Read Archive (
http://www.ncbi.nlm.nih.gov/Traces/sra/) with project number PRJNA387094. In all, 58,263,364 sequence reads were generated, of which 54,180,410 were of acceptable quality. The
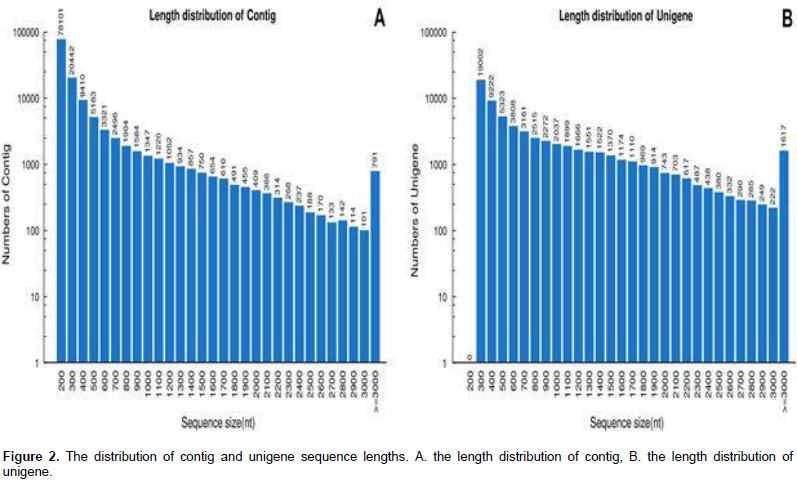
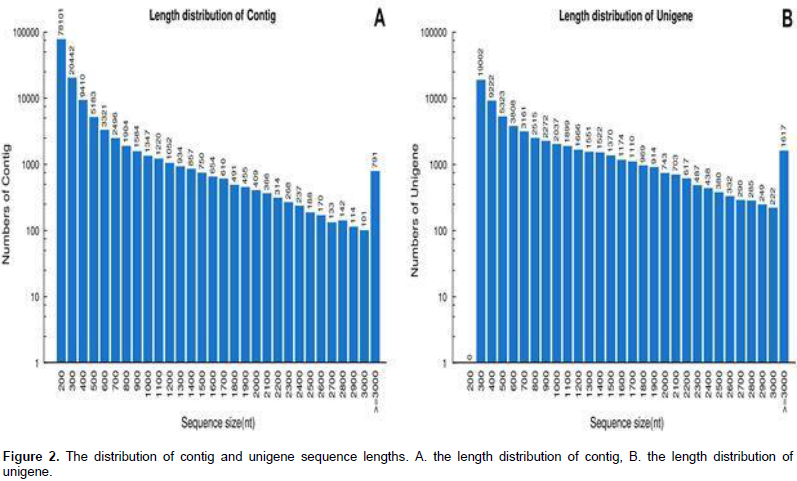
de novo data assembly yielded 134,044 contigs of mean length 347 bp (Figure 2A), and these were resolved into 65,878 unigenes, of which22,623 were clusters and 43,255 were singletons (of length at least 200 bp). The range of unigene length was from 200 bp to 11,727 bp (mean 823 bp, an N50 length of 1,357bp). Among them, 33,547 (50.9%) uni-genes were 301 to 500 bp long, 13,793 (20.9%) were 500–to 1,000 bp long, 12,918 (19.6%) were 1,000 to 2,000 bp long, and 5,620 (8.6%) were longer than 2,000 bp (Figure 2B).
Structural and functional annotation
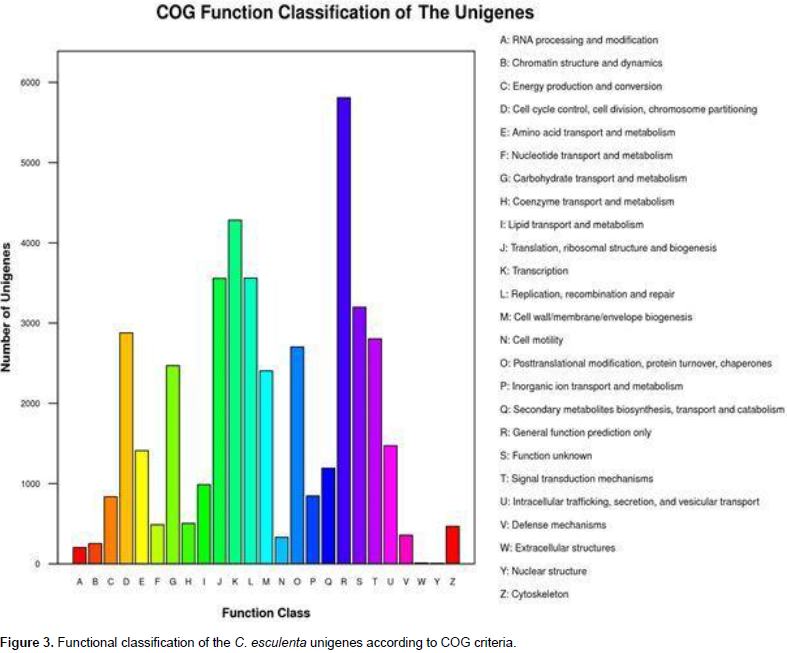
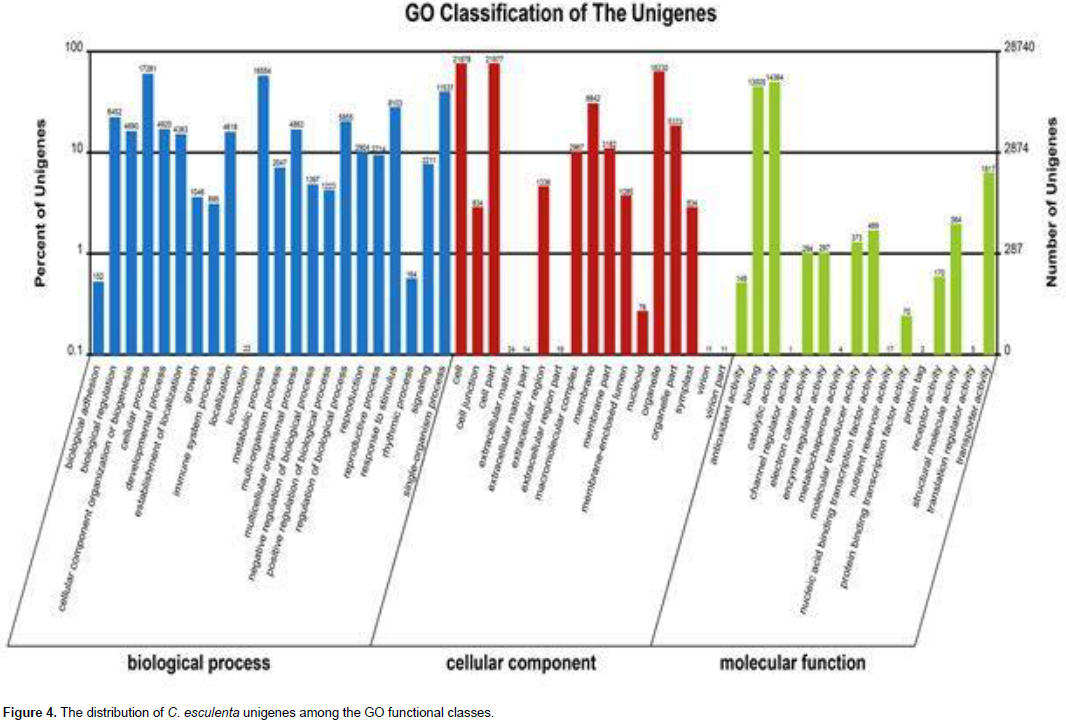
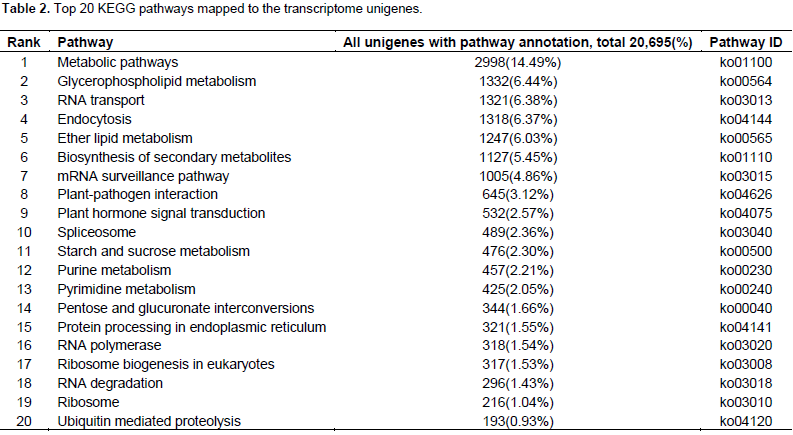
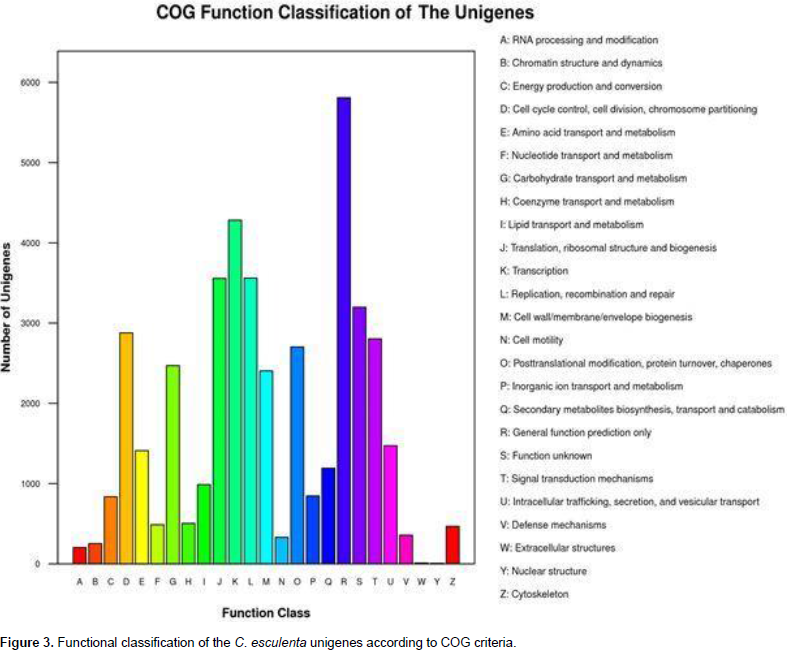
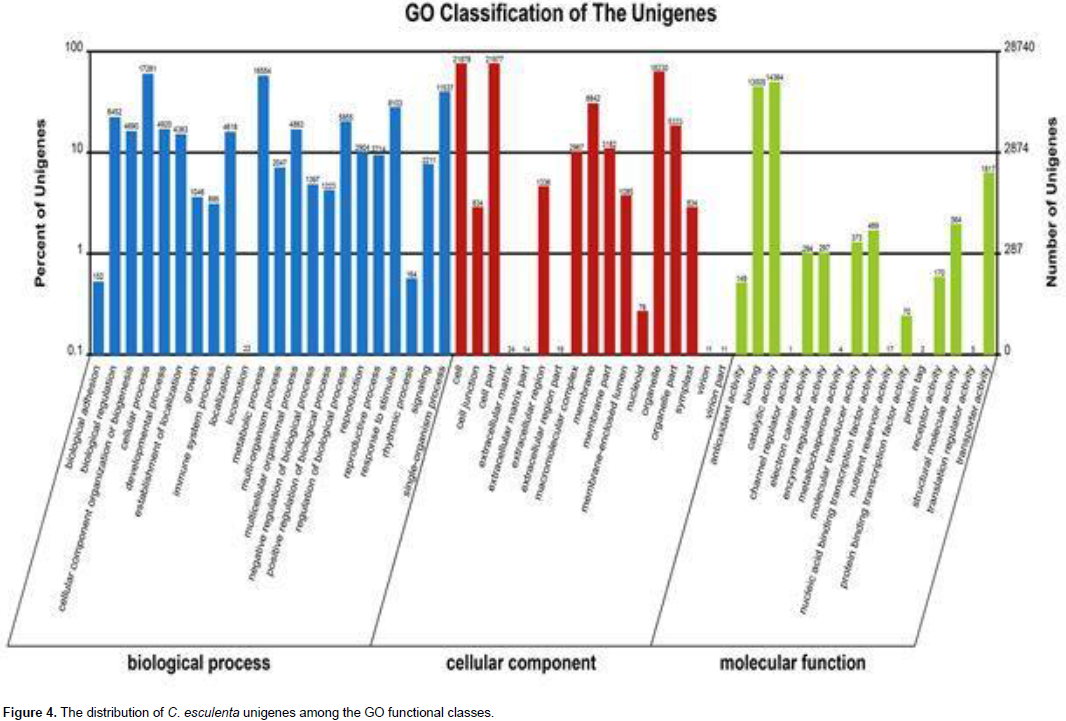
A total of 40,375 unigenes were annotated by searches against the nr, nt, UniProt, COG, GO, and KEGG databases, and shared similarity to known genes. Among them, 16,643 unigenes were identified as COG-annotated putative proteins and classified into 25 functional categories (Figure 3). The major cluster was “general function” (5,808 unigenes, 34.90%), followed by “transcription” (4,283 unigenes, 25.73%), “replication, recombination and repair” (3,561 unigenes, 21.40%) and “Translation, ribosomal structure and biogenesis” (3,557 unigenes, 21.37%). Also, 28,740 nonredundant transcripts were assigned GO terms under the three main categories as follows (Figure 4): under biological process, transcripts in cellular process (GO:0009987; 17,281 unigenes, 60.13%) and metabolic process(GO:0008152; 16,554 unigenes, 57.60%) were highly represented; under cellular component, the major terms were cell (GO:0005623; 21,978 unigenes, 76.47%) and cell part (GO:0044464; 21,977unigenes, 76.47%); and under molecular function, catalytic activity (GO:0003824; 14,364 unigenes, 49.98%) was the most dominant term, followed by binding (GO:0005488; 13,005 unigenes, 45.25%). In addition, 20,695 unigene sequences were mapped to 127 KEGG pathways. The number of unigenes inthese pathways ranged from 3 to 2,998. The top 20 pathways with the greatest number of sequences are listed in Table 2.
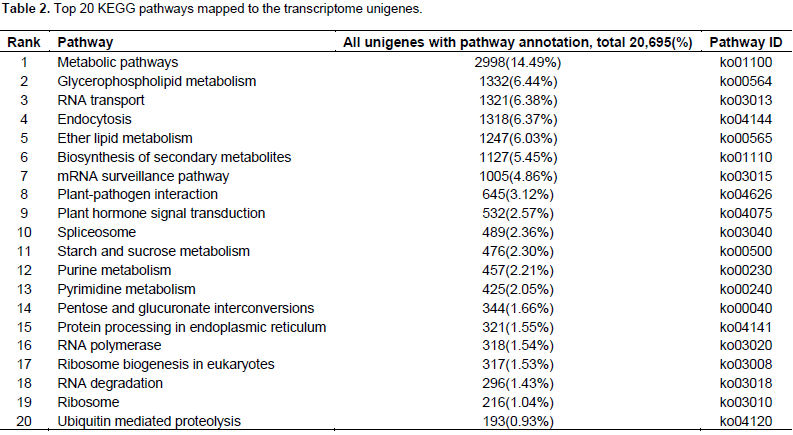
Characteristics of SSR markers
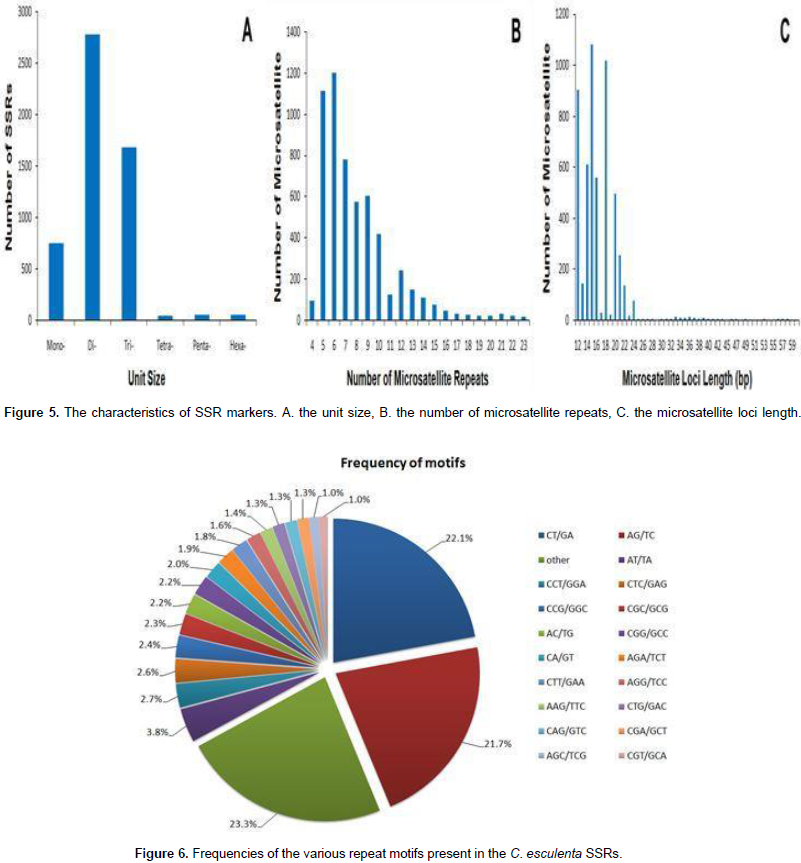
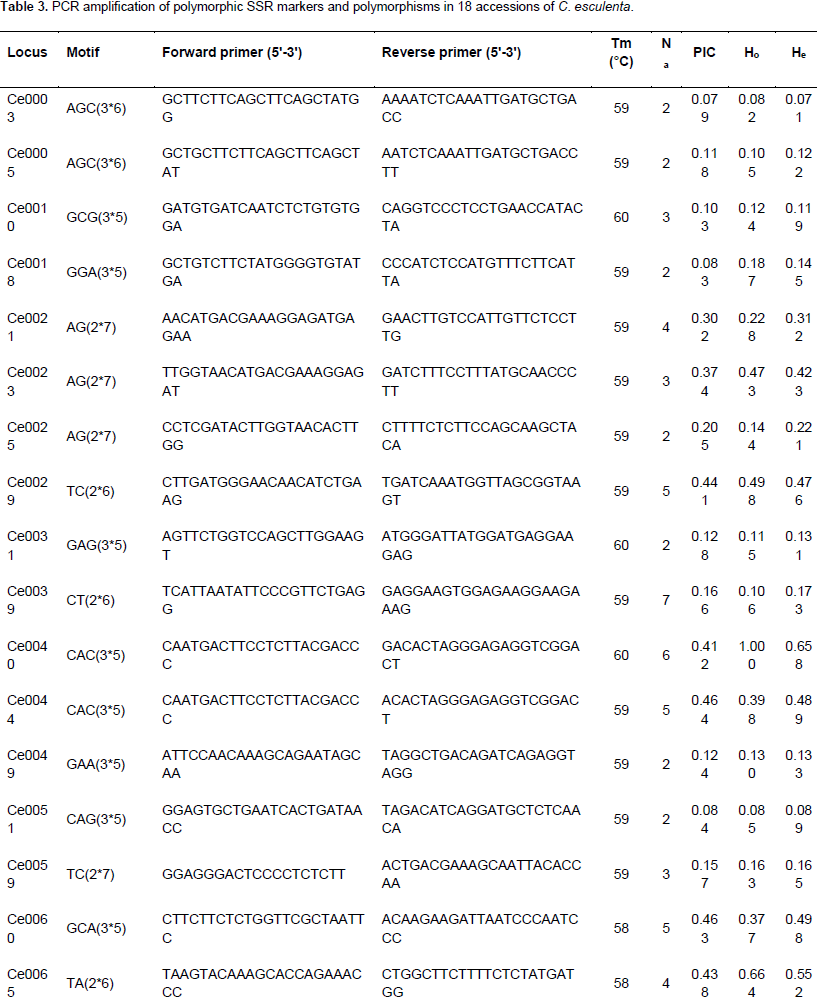
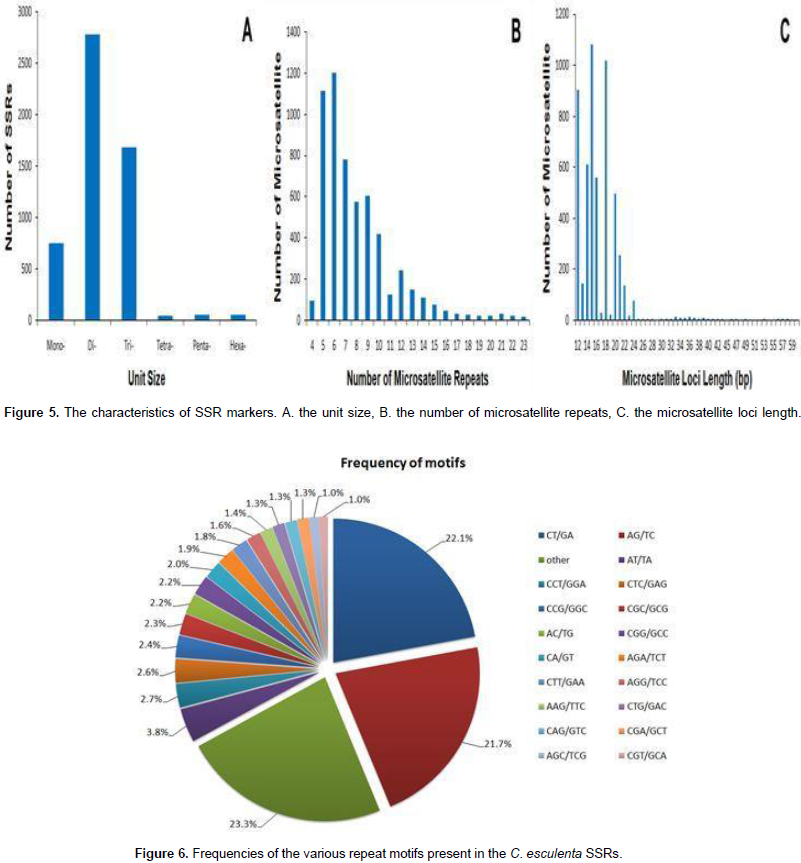
About 65,878 unigenes were used to detect potential microsatellites loci through MISA analysis, and 11,363 putative microsatellites were identified in 5,671 non-redundant unigenes, equivalent to one locus per 4.7 kb of the C. esculenta transcriptome. The most abundant repeat motifs were di-nucleotides (5,928 unigenes, 52.2%), followed by tri-nucleotides (3,491 unigenes, 30.7%), mono-nucleotide (1,629 unigenes, 14.3%), hexa-nucleotides (130 unigenes, 1.1%), penta-nucleotide (114 unigenes, 1.0%) and tetra-nucleotide (71 unigenes, 0.6%) (Figure 5). Over 180 motifs were identified, of which the most frequent were CT/GA (2,464 unigenes, 22.1%), AG/TC (2,511 unigenes, 21.7%), A/T (1,316 unigenes, 11.6%), AT/TA (431 unigenes, 3.8%), C/G (313 unigenes, 2.8%) and CCT/GGA (302 unigenes, 2.7%) (Figure 6). Based on the length of the repeat motif, the sequences were classified into two groups: Class I were hypervariable markers, consisted of SSRs ≥20 bp; Class II, or potentially variable markers were consisted of SSRs 12–20 bp of which 467 (10.7%) targeted Class I loci and the remaining ones displayed Class II loci. Almost all the sequences (95.6%) shared high homology to known genes. A further 907 putative SSRs were located among the EST sequences lodged in GenBank.
SSR markers validation
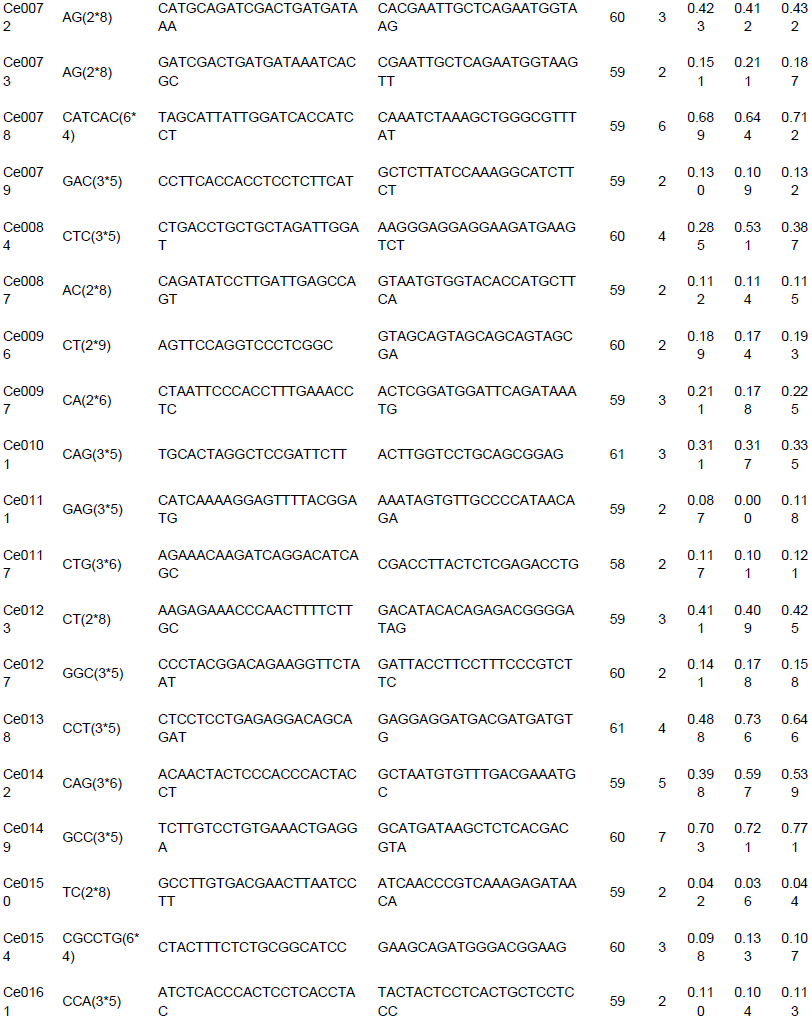
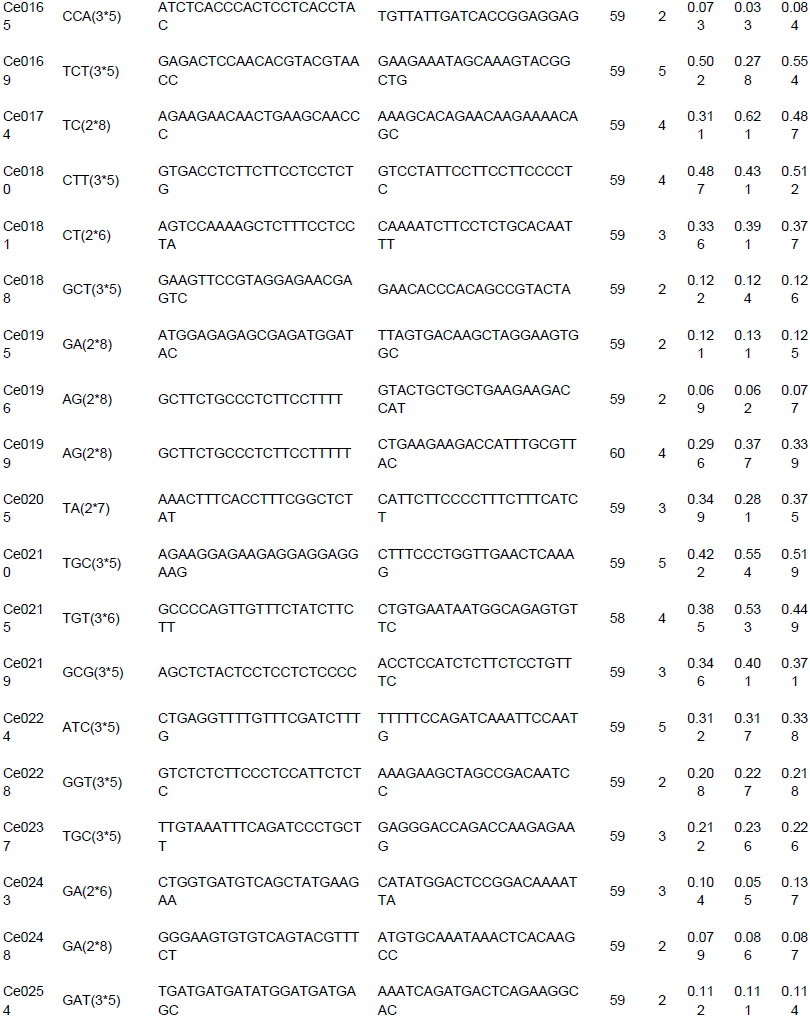
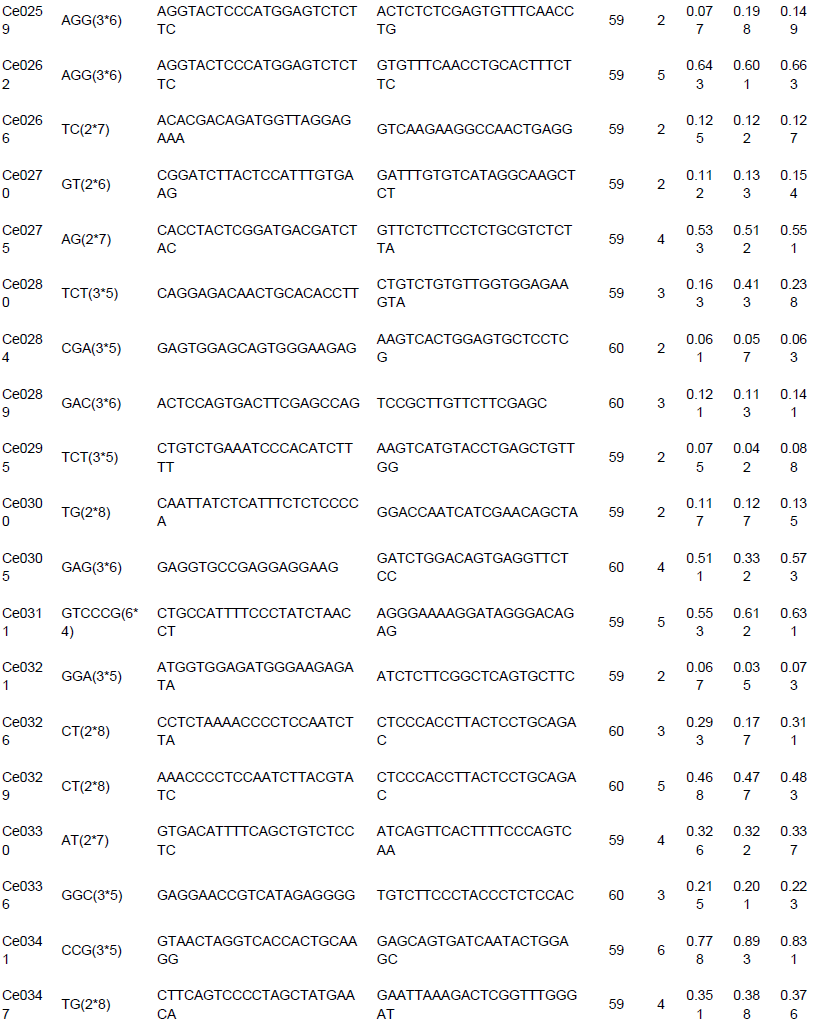
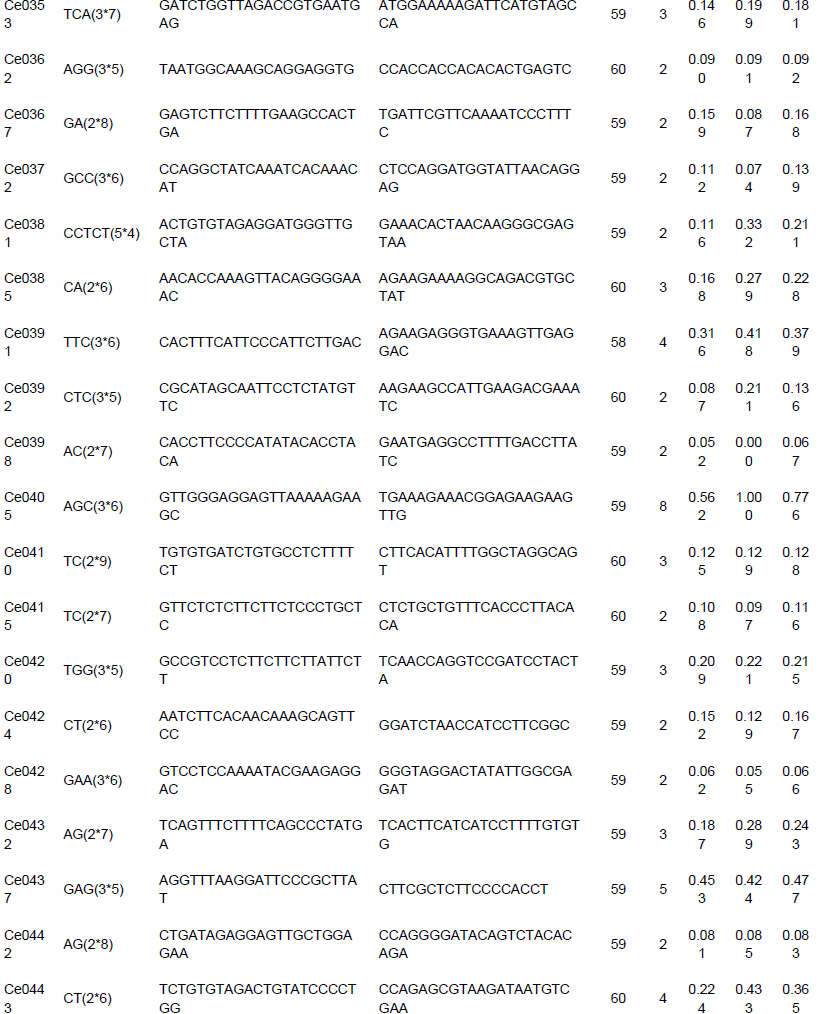
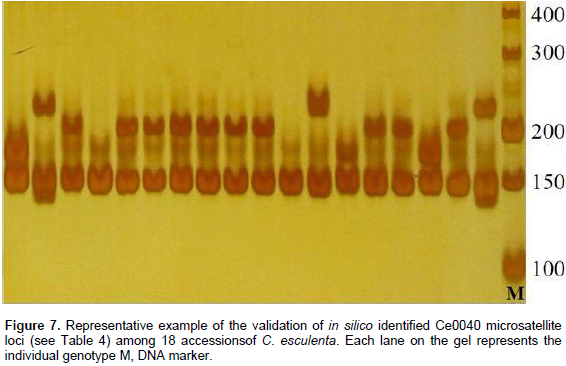
To evaluate the applicability and polymorphisms of the potential SSR markers, 150 primer pairs were randomly selected and validated through 18 accessions of C. esculenta. Also, 112 primer pairs were successfully amplified and 100 exhibited polymorphisms. A total of 316 alleles were identified, with an average of 3.16 alleles per locus. The number of alleles per locus ranged from 2 to 8. For example, the polymorphism in the Ce0040 locus (Table 3) is shown in Figure 7. Across the 100 microsatellite loci, the PIC values ranged from 0.042 to 0.778 (mean 0.245).
A comprehensive transcriptome of C. esculenta was obtained by sequencing a mixed cDNA library of leaf, stem and corm samples. The de novo data assembly yielded 134,044 contigs and resolved into 65,878 unigenes. The N50 length (1,357 bp) of the 65,878 assembled unigene sequences was much larger than those in previous studies (Liu et al., 2015). The longer N50 length meant a better quality of assembly in de novo RNA-Seq, which could benefit the identification of protein-coding genes (Namiki et al., 2012). It was attributed that the fine C. esculenta transcriptome
assembly might be due to the application of 100 bp paired-end modes for RNA-Seq, which greatly improved transcript construction and scaffolding effects (Trapnell et al., 2013; Chen et al., 2016). To predict the functions of the transcriptome sequences, the unigenes were blasted in six public databases. Total 40,375 unigenes (61.29%) were assigned at least one functional annotation with thedistribution and composition of the assigned GO terms indicating the functional distribution and evolution of conserved genes. What’s more, a large percentage of the unigenes were mapped into KEGG pathways, and most were involved in folding, sorting and degradation, transcription, translation and signal transduction pathways.
Compared with previous reports, which only identified about 32.20% in data against the four public databases, the results indicated a greater number in the annotated unigenes. In addition, results showed that although a comprehensive transcriptome of one species could be obtained by NGS, genetic and functional resources for taro are still insufficient (Nguluta et al., 2016). In the study, the large number of the C. esculenta unigenes (approximately 40%) failed to find hits in any databases, perhaps due to the lack of public sequence resources for taro or presence of non-coding transcripts among unigenes. After all, many specific functional genes in taro or that displayed low similarity to homologous genes in non-model organisms increased the difficulty to find matches in public databases (Ellegren, 2014). Traditional methods for microsatellite development mainly depended with the use of public resources, such as genetic/genomic information and EST data (Cloutier et al., 2009), and the utilization of transferable microsatellites from related species (Mathithumilan et al., 2013).
The application of NGS supplies a new and easier shortcut for SSR markers development directly from transcript sequences (Edwards et al., 2013). Here, we predicted microsatellite loci among the 65,878 assembled unigenes and 11,363 potential microsatellites had been detected among 5,671 non-redundant unigenes. The percentage of genes possessing SSR markers was about 8.61% (5,671/65,878), and the di-nucleotides repeat motifs showed the most abundance. In addition, a large number of mono-nucleotide repeat motifs were detected, which had not been discovered in the previous study of taro (You et al., 2015), while the percentage of this type in the results was about 14.3%. Although mono-nucleotide repeats were discarded for difficulty to distinguish genuine mono-nucleotide repeats from polyadenylation products, it could be an important resource for further research (Fu et al., 2013).
Among the 150 potential SSR markers, 112 loci were successfully amplified and 100 exhibited polymorphisms. This success rate (66.7%) was higher than that reported in taro EST-SSR development (You et al., 2015). Thus, the results showed that more than half of the in silico identified microsatellites and was able to be validated and provide enough number of markers for future genetic studies in taro. The unamplified loci in the study could be caused by exists of chimeric primers, primer location across splice sites, or sequences missing (Hause et al., 2016). In summary, the results provided valuable resources for future research of genetic diversity, linkage mapping, germplasm characterization and marker assisted selection in taro, which could be beneficial to breeders/geneticists and taro farmers. The transcriptomic data and microsatellite markers of taro could also be applied to the genetic researches in other species and genera of Araceae as the high transferability.
The authors have not declared any conflict of interests.
This work was supported by funding from the Natural Science Foundation of Jiangsu Province (BK20140752), National Natural Science Foundation of China (31501776) and the Fundamental Research Funds for Jiangsu Academy of agricultural sciences (ZX(15)4014).
REFERENCES
|
Ahmed I, Matthews PJ, Biggs PJ, Naeem M, Mclenachan PA, Lockhart PJ (2013). Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 13(5):929-937.
Crossref
|
|
|
|
Chaïr H, Traore RE, Duval MF, Rivallan R, Mukherjee A, Aboagye LM, Van Rensburg WJ, Andrianavalona V, De Pinheiro Carvalho MAA, Saborio F, Prana MS, Komolong B, Lawac F, Lebot V (2016). Genetic diversification and dispersal of taro (Colocasia esculenta (l.) schott). PloS One 11(6):e0157712.
Crossref
|
|
|
|
|
Chen X, Li J, Xiao S, Liu X (2016). De novo assembly and characterization of foot transcriptome and microsatellite marker development for Paphia textile. Gene 576:537-543.
Crossref
|
|
|
|
|
Cloutier S, Niu Z, Datla R, Duguid S (2009). Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor. Appl. Genet. 119:53-63.
Crossref
|
|
|
|
|
Dai HJ, Zhang YM, Sun XQ, Xue JY, Li MM, Cao MX, Shen XL, Hang YY (2016). Two-step identification of taro (Colocasia esculenta cv. Xinmaoyu) using specific psbE-petL and simple sequence repeat-sequence characterized amplified regions (SSR-SCAR) markers. Genet. Mol. Res. 15(3):gmr.15038108.
|
|
|
|
|
Das A, Das AB (2014). Karyotype analysis of ten draught resistant cultivars of Indian taro - Colocasia esculenta cv. antiquorom Schott. Nucleus (India) 57(2):113-120.
Crossref
|
|
|
|
|
Das AB, Das A, Pradhan C, Naskar SK (2015). Genotypic variations of ten Indian cultivars of Colocasia esculenta var. antiquorom Schott. evident by chromosomal and RAPD markers. Caryologia 68(1):44-54.
Crossref
|
|
|
|
|
Doungous O, Kalendar R, Adiobo A, Schulman AH (2015). Retrotransposon molecular markers resolve cocoyam (Xanthosoma sagittifolium) and taro (Colocasia esculenta) by type and variety. Euphytica 206(2):541-554.
Crossref
|
|
|
|
|
Edwards D, Batley J, Snowdon RJ (2013). Accessing complex crop genomes with next-generation sequencing. Theor. Appl. Genet. 126:1-11.
Crossref
|
|
|
|
|
Ellegren H (2014). Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 29:51-63.
Crossref
|
|
|
|
|
Fu N, Wang Q, Shen HL (2013). De novo assembly, gene annotation and marker development using Illumina paired-end transcriptome sequences in celery (Apium graveolens L.). PloS One 8:e57686.
Crossref
|
|
|
|
|
Hause RJ, Pritchard CC, Shendure J, Salipante SJ (2016). Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 11:1342-1350.
Crossref
|
|
|
|
|
He J, Zhao X, Laroche A, Lu ZX, Liu H, Li Z (2014). Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front Plant Sci. 5:484.
Crossref
|
|
|
|
|
Hunt HV, Moots HM, Matthews PJ (2013). Genetic data confirms field evidence for natural breeding in a wild taro population (Colocasia esculenta) in northern Queensland, Australia. Genet. Resour. Crop Evol. 60:1695-1707.
Crossref
|
|
|
|
|
Liu HB, You YN, Zheng XF, Diao Y, Huang XF, Hu ZY (2015). Deep sequencing of the Colocasia esculenta transcriptome revealed candidate genes for major metabolic pathways of starch synthesis. S. Afr. J. Bot. 97:101-106.
Crossref
|
|
|
|
|
Liu K, Muse SV (2005). PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128-2129.
Crossref
|
|
|
|
|
Mathithumilan B, Kadam NN, Biradar J, Reddy SH, Ankaiah M, Narayanan MJ, Makarla U, Khurana P, Sreeman SM (2013). Development and characterization of microsatellite markers for Morus spp. and assessment of their transferability to other closely related species. BMC Plant Biol. 13:194.
Crossref
|
|
|
|
|
Namiki T, Hachiya T, Tanaka H, Sakakibara Y (2012). MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 40:e155.
Crossref
|
|
|
|
|
Nath VS, Sankar MSA, Hegde VM, Jeeva ML, Misra RS, Veena SS, Raj M (2014). Analysis of genetic diversity in Phytophthora colocasiae causing leaf blight of taro (Colocasia esculenta) using AFLP and RAPD markers. Ann. Microbiol. 64(1):185-197.
Crossref
|
|
|
|
|
Nguluta M, Adebola P, Pillay M (2016). Genetic diversity analysis in South African taro (Colocasia esculenta) accessions using molecular tools. Int. J. Genet. Mol. Biol. 8:18-24.
Crossref
|
|
|
|
|
Oliveira LSS, Harrington TC, Ferreira MA, Freitas RG, Alfenas AC (2017). Populations of Ceratocystis fimbriata on Colocasia esculenta and other hosts in the Mata Atlântica region in Brazil. Plant Pathol.
|
|
|
|
|
Ramanatha RV, Matthews PJ, Eyzaguirre PB, Hunter D (2010). The global diversity of taro: ethnobotany and conservation. Biodiversity International, Rome. Available at:
View.
|
|
|
|
|
Shete S, Tiwari H, Elston RC (2000). On estimating the heterozygosity and polymorphism information content value. Theor. Popul. Biol. 57:265-271.
Crossref
|
|
|
|
|
Soulard L, Letourmy P, Cao TV, Lawac F, Chaïr H, Lebot V (2016). Evaluation of vegetative growth, yield and quality related traits in taro (Colocasia esculenta [L.] schott). Crop Sci. 56(3):976-989.
Crossref
|
|
|
|
|
Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31:46-53.
Crossref
|
|
|
|
|
Vandenbroucke H, Mournet P, Vignes H, Chaïr H, Malapa R, Duval MF, Lebot V (2016). Somaclonal variants of taro (Colocasia esculenta Schott) and yam (Dioscorea alata L.) are incorporated into farmers' varietal portfolios in Vanuatu. Genet. Resour. Crop Evol. 63(3):495-511.
Crossref
|
|
|
|
|
Waples RK, Larson WA, Waples RS (2016). Estimating contemporary effective population size in non-model species using linkage disequilibrium across thousands of loci. Heredity 117:233-240.
Crossref
|
|
|
|
|
Yeh FC, Yang R, Boyle TJ, Ye Z, Xiyan JM (2000). PopGene32, Microsoft Windows-based freewarefor population genetic analysis, version 1.32. Molecular Biology Biotechnology Center, University of Alberta, Edmonton, Alberta, Canada.
|
|
|
|
|
You YN, Liu DC, Liu HB, Zheng XF, Diao Y, Huang XF, Hu ZY (2015). Development and characterisation of EST-SSR markers by transcriptome sequencing in taro (Colocasia esculenta (L.) Schoot). Mol. Breed. 35:134.
Crossref
|
|