Herbal medicine has been around since human civilization and continues to play a vital role in health care (Deepika and Yash, 2013). Last decade, several molecules used as drugs were isolated from natural resources on the base of traditional uses (Murugan and Mohan, 2011). Majority of resources are made up of medicinal plants which possess compounds (secondary metabolites). These compounds are diverse and are responsible several pharmacological properties including antioxidant, anti-inflammatory, antibacterial, antidiabetic, hepatoprotective, etc (Wadood et al., 2013).
Inflammation is the living organism’s response to any aggression (pathogens, injury). During inflammation process, inflammatory mediators such as prostaglandins, leukotrienes, cytokines (TNFα, IL1β, IL6) and reactive oxygen species (ROS) are released by cells activation. Prostaglandins and leukotrienes are respectively produced by cyclooxygenases (COX) and lipoxygenase (LOX) activation (Hunter, 2012). Inflammatory mediators and redox status participate in a significant disease process in both acute and chronic inflammatory states. This involves major cascades of release of inflammatory mediators, which are generally associated with oxidative damage to cellular constituents (Sharma et al., 2019).
Oxidative stress and ROS are mainly associated with the pathophysiology of major chronic diseases such as inflammation, cancer, atherosclerosis, diabetes, and arthritis (Chikara et al., 2018). Inflammatory disorders are currently treated with steroidal / nonsteroidal anti-inflammatory drugs that induce side effects (gastric, renal, and cardiovascular disorders) (Jordan and White, 2001).
Medicinal plants used to treat inflammation are a natural source of discovery of anti-inflammatory agents with fewer side effects. According to WHO, around 80% of population in developing countries use plants for their primary health care. However, most of medicinal plants are used world-wide without scientific data about their possible toxic effects. World Health Organization has recommended that medicinal plants used to treat human diseases be the subject of further scientific investigation on their side effects (WHO, 2008; Koriem et al., 2019). So, plants should be studied in order to better understand their efficacy, properties and safety (Owolabi et al., 2007; Koriem et al., 2019).
Cocos nucifera L., belonging to Arecaceae family (palm family) known as coconut and Carica papaya L. derived from Caricaceae family are two medicinal plants cultivated currently in the hot and humid countries (America, Africa, India, Brazil). All of their constituents have various benefits to the human body through effects on inflammation, nociception, oxidative stress, fever, dysentery, tumor (Maisarah et al., 2014; Lima et al., 2015). The aim of this study was to determine the phytochemical composition and to assess in vitro the antioxidant, anti-inflammatory properties and safety of methanolic and aqueous extracts from the roots of C. nucifera L. and C. papaya L.
Chemicals
Trolox was purchased from Fluke, France. Iron dichloride, hydrochloric acid, indomethacin, hydrogen peroxide, ABTS [2,2'-azinobis (3-ethyl benzoin-6- sulphonate)], trichloroacetic acid, aluminum trichloride, ammonia, by Prolabo (Paris, France). Folin Ciocalteu reagent, gallic acid, quercetin, ferric chloride, DPPH (2, 2-diphenyl-1-picrylhydrazyl), trichloroacetic acid, hydrochloric acid, potassium persulfate, ascorbic acid, ketamine, potassium hexacyanoferrate, 2-thiobarbituric acid, sodium tetraborate, boric acid, zileuton, linoleic acid, tween 20, and lipoxygenase (type I-B) enzyme were purchased from Sigma® (St Louis, USA). COX-1 and human COX-2, Screening Kit (Item No. 560131) and sPLA2 (Item No. 765001) were manufactured by Cayman Chemical Co. (MI, USA). All solvents used were of analytical grade.
Plant materials and extraction
C. papaya L. (Caricaceae) roots were harvested in September 2018 around Dedougou in the region of the mouhoun loop located 250 km to the capital of Burkina Faso (N 12°46’44.6; W 003°44’91.4). A sample was identified and authenticated at the Plant Biology and Ecology Laboratory of University Joseph KI-ZERBO. The voucher specimen was deposited under number T4316. C. nucifera L. (Arecaceae) roots were provided by a tradipractician and were also authenticated at the same laboratory. The roots of two plants were rinsed with running water, and dried under ventilation out of the dust and light; then, they were powdered by Gladiator Est. 1931 Type BN 1 Mach. 40461 1083.
A quantity of 50 g of each powder was macerated in water (500 ml) for 24 h. After filtration, the extracts were centrifuged and lyophilized to obtain dried extract. A methanolic maceration was realized using the same method but after filtration with whatman’s filter paper, the extracts were concentrated with rotary vacuum evaporator and kept in an oven until complete evaporation of solvent. The extracts obtained were kept cool for further investigations.
Animals and ethical approval
Female NMRI mice weighing between 20 - 35 g from the animal’s house of Institute of Health Sciences Research were used for toxicological assays of extracts. The animals were maintained at laboratory breeding conditions (temperature of 20 - 25 °C, 12 h light/12 h dark cycle, and humidity of 60%). They were fed with standard laboratory pellet (29% protein) and running water. The laboratory experimentation was carried out according to the experimental protocols already validated by the Institute of Health Sciences Research laboratories and meeting the international standards in this field (guidelines established by the European Union on the protection of animals, CCE Conseil 86/609).
Phytochemical screening
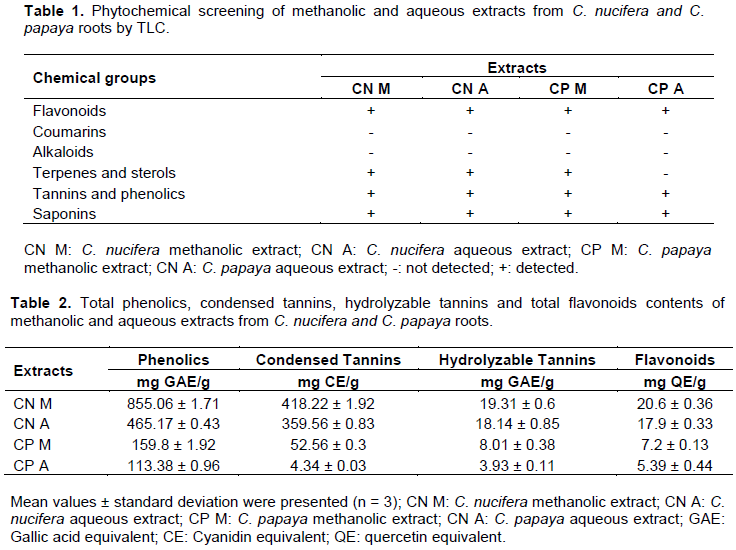
The phytochemical screening of extracts from plants was carried out by thin layer chromatography (TLC, 60 F254, 10 x 5 cm, 10 x 20cm glass support, Merck) in accordance with literature methods (Ladigina et al., 1983; Santiago and Strobel, 2013). Each dry extract was solubilized in methanol and deposited on the plate for the evolution of the chromatogram. The purpose of this test was to screen large chemical groups like sterols, triterpenes, flavonoids, tannins, alkaloids, coumarins which are secondary metabolites with several pharmaceutical properties.
Specific reagents were used to reveal these groups of compounds: Dragendorff reagent for alkaloids; 5% ethanol FeCl3 reagent for tannins and phenolics; Neu's reagent for flavonoids; Sulfuric vanillin reagent for terpenes and sterols; Anysaldehyde reagent for saponosides; and 5% methanolic KOH reagent for coumarins.
Phytochemical composition
Total phenolics content
The total phenolics of aqueous and methanolic extracts were carried out according to method described by Singleton et al. (1999). To do this, 25 µl of each extract (100 µg/ml) was mixed with 125 µl of Folin Ciocalteu Reagent (FCR 0.2 N). After 5 min at ambient temperature, 100 µl of sodium carbonate solution (75 g/L) was added. The mixtures were incubated during 1 h at room temperature and the absorbances were measured (Spectrophotometer UV, Epoch Biotek Instruments, U.S.A.) at 760 nm against blank. A standard calibration curve was calculated by using Gallic acid. The mixture made in triplicate and the results were expressed in mg of Gallic acid equivalent per g of extract (mg GAE/g).
Tannins content
Condensed tannins
The method used to determine the condensed tannins content is that described by Swain and Hillis (1959). 1 ml of extract (5 mg/ml) was added to 2 ml of vanillin 1% (1 g of vanillin and 100 ml of 70 % sulfuric acid). After 15 min incubation in water bath at 20°C, the absorbance of the mixture was measured (spectrophotometer UV, Shimadzu) at 500 nm. The condensed tannins content T (%) was determined using the following formula:
T (%) = 5.2×10-2 × (A x V / P)
5.2×10-2 = constant in equivalence of cyanidin, A = absorbance, V = extract volume and P = extract weight.
The condensed tannins content of the samples was determined in triplicates and the results were converted to mg of cyanidin equivalent (CE) / g dry extract.
Hydrolyzable tannins
The hydrolyzable tannins were performed to Mole and Waterman (1987) method. 1 ml of each extract (5 mg/ml) was mixed to 3.5 ml of the reagent (ferric chloride FeCl3 10-2 M in hydrochloric acid HCl 10-3 M). The absorbance of the mixture was measured (spectrophotometer UV, Shimadzu) at 660 nm after 15 s incubation. The hydrolysable tannins content T (%) was determined according to the formula below:
T (%) = (A × PM × V ×FD) / ε mole × P
A = absorbance, PM = weight of gallic acid (170.12 g/mol), V = volume of extract, FD = dilution factor, ε mole = 2169 (constant in equivalence of gallic acid), P = extract weight
The hydrolysable tannins content of the samples was determined in triplicates and the results were converted to mg of gallic acid equivalent (GAE) / g dry extract.
Flavonoids content
The total flavonoïds of the extract were measured by aluminium chloride (AlCl3) colorimetric assay (Arvouet-Grand et al., 1994). Each extract (1 mg) was dissolved in methanol (1 mL). 100 µL of AlCl3 solution (2 % in methanol) was added to equal volumes of extracts. After shaking, the mixture was incubated for 10 min, and the absorbance was measured at 415 nm with spectrophotometer (Spectrophotometer UV, Epoch Biotek Instruments, U.S.A.) against blank. The blank was composed of 100 µL methanol and 100 µL of each extract. Quercetin was used to produce the standard curve. The flavonoïd content of extracts was determined in triplicates and the results were expressed as mg of quercetin equivalent (QE)/g dry extract.
Anti-inflammatory activity
Phospholipase A2 inhibition assay
The sPLA2 inhibition test from Cayman Laboratories allows the screening of sPLA2 inhibitors (Type V). The assay was determined with the method described by Cayman Chemical Co. (MI, USA) in the catalog No. 765001. The assay was done in triplicate using 96-wells microplate. The absorbances were read (Agilent 8453) at 405 nm against a blank that had not received the enzyme. Ascorbic acid was used as reference compound and sPLA2 inhibition percentage per 100 μg/mL (final concentration in the wells) was calculated with the formula:
% Inhibition = [(AEA - AIA) / AEA] × 100
Where, AEA: Activity enzyme test absorbance; AIA: Activity inhibition test Absorbance
Cyclooxygenases 1 and 2 inhibition assay
The cyclooxygenases Cox-1 (ovine) and Cox-2 (ovine recombinant) inhibition assay was carried out using a commercial colorimetric inhibitor test kit (Catalog No. 560131, Cayman Chemical Company, U.S.A). The tests were carried out following manufacture’s instructions. Analysis was performed spectrophotometrically Epoch (Bioteck Instruments, U.S.A) at wavelength of 590 nm against blank. Indomethacin and ascorbic acid were used as reference compound and percentage of inhibition induced by 100 μg/mL was given by the formula:
% Inhibition = [(AEA - AIA) / AEA] × 100
Where, AEA: Activity enzyme test absorbance, AIA: Activity inhibition test Absorbance
Lipoxygenase inhibition assay
The inhibition of lipoxygenase was determined by Malterud and Rydland (2000)’s method. For this purpose, 3.75 μl of the extracts
at concentration of 8 mg/mL was mixed with 146.25 µl of lipoxygenase solution (820.51 U/ml) prepared in boric acid buffer (pH 9, 0.2 M). The mixture was incubated at ambient temperature during 3 min and 150 μl of 1.25 mM of linoleic acid (substrate) was added. Spectrophotometer (Epoch Biotek Instruments, U.S.A.) was used to record the absorbances for 3 min at 234 nm. The tests were performed in triplicate and zileuton was used as reference compound. The percentage of lipoxygenase inhibition was calculated using the formula:
% Inhibition = (Vb − Vs) / Vb ×100
Vb: Enzymatic activity without inhibitor; Vs Sample: Enzymatic activity with sample/reference compound
Antioxidant activity
Lipid peroxidation inhibitory test (LPO)
The inhibitory capacity of lipid peroxidation activity was evaluated with rat liver using Sombié’s method (Sombié et al., 2011). 0.2 ml of extracts or positive control (ascorbic acid) at a concentration of 1.5 mg / ml was mixed with 1 mL of liver homogenate in 10% phosphate buffered saline (PBS) buffer (pH 7.4), 50 μl of FeCl2 (0.5 mM) and then 50 μl of H2O2 (0.5 mM). After one-hour incubation at room temperature, 1 ml of trichloroacetic acid (15 %) and 1 ml of 2-thiobarbituric acid (0.67%) were added. The mixture was incubated for 15 min in boiling water and centrifuged (2000 rpm for 10 min). The absorbances were read with a spectrophotometer (Epoch Biotek Instruments, U.S.A.) at 532 nm against control (without extract). All of these measurements were carried out in triplicate. The percentage of inhibition induced by 100 μg/ml was calculated as follows:
%Inhibition = (Ab – Ae) /Ab × 100
Ab: absorbance of control; Ae: absorbance of extracts/ reference compound
FRAP (ferric reducing antioxidant power) test
The ability of extracts to reduce Fe3+ ion to Fe2+ ion was evaluated using the method described by Hinneburg et al. (2006). To 500 µl of each extract (1 mg/ml), were added 1.25 ml of phosphate buffer (pH 6.6, 0.2 M), and then 1.25 ml of potassium hexacyanoferrate solution [K3Fe(CN)6] (1% in water). After 30 min incubation in a water bath at 50°C, 1.25 ml of trichloroacetic acid (10 %) was added and the mixture was centrifuged (2000 rpm) for 10 min. 625 µl of the supernatant was mixed with 625 µl of distilled water and then 125 µl of freshly prepared 0.1% FeCl3 in water. A blank without sample is prepared under the same conditions. The absorbances were read at 700 nm with spectrophotometer (Epoch Biotek Instruments, U.S.A) against a standard curve of ascorbic acid. The potential of extracts to reduce iron (III) to iron (II) was expressed in millimole Ascorbic Acid Equivalent per gram of dry extract (mmol AAE/g).
DPPH (2,2-diphenyl-1-picrylhydrazyl) test
The capacity of extracts to scavenge DPPH radical was assayed as described by Velazquez et al. (2003). A cascade dilution of the extract and reference substances (Trolox and ascorbic acid) was performed from a concentration of 1 mg/ml. For this purpose, 200 µl of 4% DPPH solution (in methanol) freshly prepared was mixed with 100 μl of each dilution in the 96-wells microplate. The mixture was incubated for 30 min at ambient temperature. The absorbances were read (Epoch Biotek Instruments, U.S.A.) at 517 nm against a blank (methanol). The percent inhibition was calculated as follows:
% Inhibition = (Ab – Ae) /Ab × 100
Ab: absorbance of blank; Ae: absorbance of extract/reference compound
ABTS (2, 2’-azinobis- [3-ethylbenzothiazoline-6-sulfonic acid]) test
ABTS radical cation scavenging ability of extracts was determined according to Re et al. (1999)’s procedure. On the eve of the test, a stock solution of ABTS (7 mM) was prepared with 2.45 mM of potassium persulfate (K2S2O8) and the mixture was stored in the room without light for 12 to 16 h. A cascade dilution range of the extracts and reference substances (Trolox and ascorbic acid) were realized from a concentration of 1 mg/ml. 20 µl of each dilution was mixed with 200 µL of the ABTS solution diluted in ethanol in the 96-wells microplate. The absorbances were read against blank (ethanol) on a spectrophotometer (Epoch Biotek Instruments, U.S.A.) at 734 nm, after 30 min of incubation in the dark at room temperature. The ABTS radical inhibition was determined by the formula below:
% Inhibition = [(Ab – Ae) /Ab] × 100
Ab: absorbance of blank; Ae: absorbance of extract/reference compound
Acute toxicity test
The acute toxicity study was conducted according to acute toxic class method of the Organization for Economic Cooperation and Development (OECD, 2001) test guideline 423. The test was carried out twice.
Administration of extracts
Before the experiment, all the female mice were weighed, marked, and grouped randomly into five batches with three mice / group. After four hours fasting with access to running water, the control group received distilled water at a dose of 10 ml/ kg, the aqueous and methanolic extracts from Carica papaya were administered to the animals of batch 2 and 3 respectively; and the aqueous and methanolic extracts from Cocos nucifera were administered to the animals of batch 4 and 5 respectively. All of the extracts were administered to the animals at a single dose of 2000 mg/kg of body weight by oral gavage.
Daily observations
After oral gavage, the animals were observed for 2 h while animals were monitored individually and next, they were fed. All the animals were inspected individually with particular attention for fourteen days in order to detect any sign of toxicity namely general behavioral in eyes, skin, activeness, touch, and movement changes including number of deaths.
Statistical analysis
The data were expressed as Mean ± Standard Error of Mean (SEM). The statistical analysis was carried out according to one-way ANOVA analysis followed by Dunnett’s test compared to the control and between methanolic and aqueous extract on Graph Pad Prism software version 6.0. The level of significance was accepted at p < 0.05.
Phytochemical study
Phytochemical screening by TLC of C. nucifera and C. papaya roots extracts revealed the presence of secondary metabolites like flavonoids, saponins, tannins and phenolics and absence of coumarins and alkaloids (Table 1). Carica papaya methanolic extract contains sterols and triterpenes while aqueous extracts do not.
The total phenolics, condensed tannins, hydrolysable tannins and total flavonoids contents of methanolic and aqueous extracts from the plants are presented in Table 2. The methanolic extracts presented the highest total content of phenolics, flavonoids and tannins than water extracts. It has also been reported that the extraction of phenolics from the plant samples are influenced by the nature of solvent (Younus et al., 2019). This could be explained by the efficiency of methanol (a very polar solvent) in the degradation of cell walls which have a non-polar character and cause the release of polyphenols from cells. Also, there is an enzyme that degrades polyphenols in aqueous extracts called polyphenol oxidase which may be responsible for the decrease in the activity of the aqueous extraction. In methanol extracts, this enzyme would be absent (Lapornik et al., 2005; Tiwari et al., 2011). Indeed, the phenolics, flavonoids and tannins were well documented as valuable antioxidants and anti-inflammatory (Sharma et al., 2019).

Antioxidant activity
DPPH radical scavenging, ABTS+ radical cation decolorization, ferric ion reduction and lipid peroxidation inhibition in rat liver assays were used to assess the antioxidant activity. Indeed, Reddy et al. (2012) specified that it is necessary to realize more than one antioxidant method to take into account the different antioxidant modes of action. The extracts exhibited antioxidant activity with the best activities recorded by methanolic extracts indicated in Table 3. The methanolic extract from C. nucifera demonstrated interesting results in ABTS and DPPH assays were highly comparable to Trolox as reference. The results obtained showed that the extract had a capacity to reduce the ferric ion to ferrous ion. In this test, antioxidant electron donation leads to the neutralization of the free radical (Moualek et al., 2016). The in vitro inhibition percentage on lipid peroxidation in rat liver of methanolic extracts from both plants at 100 µg/ml was greater than 50%. However, the percentage of ascorbic acid (94.95 ± 0.94) was better than all of the extracts. In general, C.nucifera showed better antioxidant power compared to C. papaya for the four methods used with the exception of aqueous extracts in the lipid peroxidation test. Peroxidation of lipids disturbs the integrity of cell membranes and lead to rearrangement of membrane structure (Ozougwu, 2016). Inhibition of extracts against lipid peroxidation suggests that extracts protect cell membrane. It has been reported in the literature that free radical plays a crucial role in the pathogenesis of several diseases including inflammation, pulmonary, cancer, rheumatoid, diabetes, cardiovascular diseases, atherosclerosis, hypertension, ischemia/ reperfusion injury (Valko et al., 2007; Reuter et al., 2010). Antioxidants are substances that prevent various pathologic changes in living cell by protecting oxidation of its major constituents (proteins, lipids, carbohydrates and DNA) (Moualek et al., 2016). Phenolic compounds are considered the most antioxidant metabolites from plants; and these compounds have the ability to give hydrogen or electrons (Koolen et al., 2013; Da Silva Santos et al., 2020).
Inhibitor effect extracts against enzymes
The results of the proinflammatory enzymes inhibition tests are presented in Table 4. These results showed that methanolic extract of C. nucifera presented the high inhibition activity of 15-lipoxygenase. However, Zileuton (reference compound) exhibited better IC50 in comparison to the extracts. C. papaya methanolic extracts were significant highly comparable with Indomethacin and ascorbic acid used as reference compounds, respectively in COX-2 and PLA2 inhibition. Inflammation is the part of biological reaction of vascular tissues to external harmful stimuli, such as pathogens, damaged cells, or irritants (Das et al., 2014). Overproduction of reactive oxygen species (ROS) during inflammatory process induces cytokines (TNFα, IL1β, IL6) release and pro-inflammatory enzymes activation (Phospholipase, cyclooxygenases, lipoxygenase) (Manouze et al., 2017). Most treatments for inflammatory diseases use non-steroidal anti-inflammatory drugs (NSAIDs).
The extracts have more affinity to inhibit the activity of cyclooxygenase 2 than that of COX-1. According to Bacchi et al. (2012), the extracts can be classified in the 2nd group of NSAIDs (capacity to inhibit COX-1 and COX-2 with a preferential selectivity toward COX-2). It is generally thought that their principal mechanism of action is the inhibition of cyclooxygenase (COX-2), the enzyme responsible for biosynthesing the prostaglandins and thromboxane (Jordan and White, 2001). However, this class of drug contains many side effects (Scheiman, 2016). It seems that simultaneously inhibiting COX and LOX, and therefore decreasing the production of leukotrienes and prostaglandins may offer clinically relevant advantages over COX inhibition (Bacchi et al., 2012). The present study showed that the extracts of Cocos nucifera and Carica papaya roots inhibited the activity of 15-LOX, sPLA2, COX-1, and COX-2, the key enzymes in the formation of eicosanoids (inflammation mediators) from arachidonic acid. The flavonoids, saponins and tannins might be responsible in part for the observed anti-inflammatory effects (Das et al., 2014; Kamau et al., 2016).
Acute oral toxicity
The results of acute toxicity study concerning mortality rate of extracts are presented in Table 5. Acute oral toxicity evaluation reported that no mortality was observed in mice with single dose of 2000 mg / kg right through the 14 days experiment. None of the extracts produced notable changes in behavior during the time of observation. The same observation was made in both steps of the study. These results suggest that aqueous and methanolic extracts from C. nucifera and C. papaya were classified in class 5 of toxicity and estimated the median lethal dose LD50 at 5000 mg / kg, according to acute toxicity class method of OECD guideline 423 (OECD, 2001). The extracts were classified to belong to substances with a low acute oral toxicity according to the Globally Harmonized System of Classification and Labeling of Chemicals of the United Nations (ONU, 2017).