Full Length Research Paper

ABSTRACT
Fungal diversity in agro-ecosystems is influenced by various factors related to soil and crop management practices. However, due to the complexity in fungal cultivation, only a limited number has been extensively studied. In this study, amplicon sequencing of the Internal Transcribed Spacer (ITS) region was used to explore their diversity and composition within long-term farming system comparison trials at Chuka and Thika in Kenya. Sequences were grouped into operational taxonomic units (OTUs) at 97% similarity and taxonomy assigned via BLASTn against UNITE ITS database and a curated database derived from GreenGenes, RDPII and NCBI. Statistical analyses were done using Vegan package in R. A total of 1,002,188 high quality sequences were obtained and assigned to 1,128 OTUs; they were further classified into eight phyla including Ascomycota, Basidiomycota, Chytridiomycota, Glomeromycota, Calcarisporiellomycota, Kickxellomycota, Mortierellomycota and unassigned fungal phyla. Ascomycota was abundant in conventional systems at Chuka site while Basidiomycota and Chytridiomycota were dominant in conventional systems in both sites. Kickxellomycota and Calcarisporiellomycota phyla were present in all organic systems in both sites. Conventional farming systems showed a higher species abundance and diversity compared to organic farming systems due to integration of organic and inorganic inputs.
Key words: Long-term farming systems, fungi, internal transcribed spacer (ITS), diversity, Illumina sequencing.
INTRODUCTION
MATERIALS AND METHODS
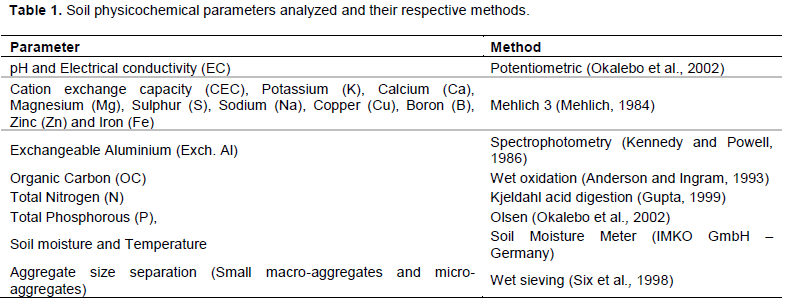
RESULTS
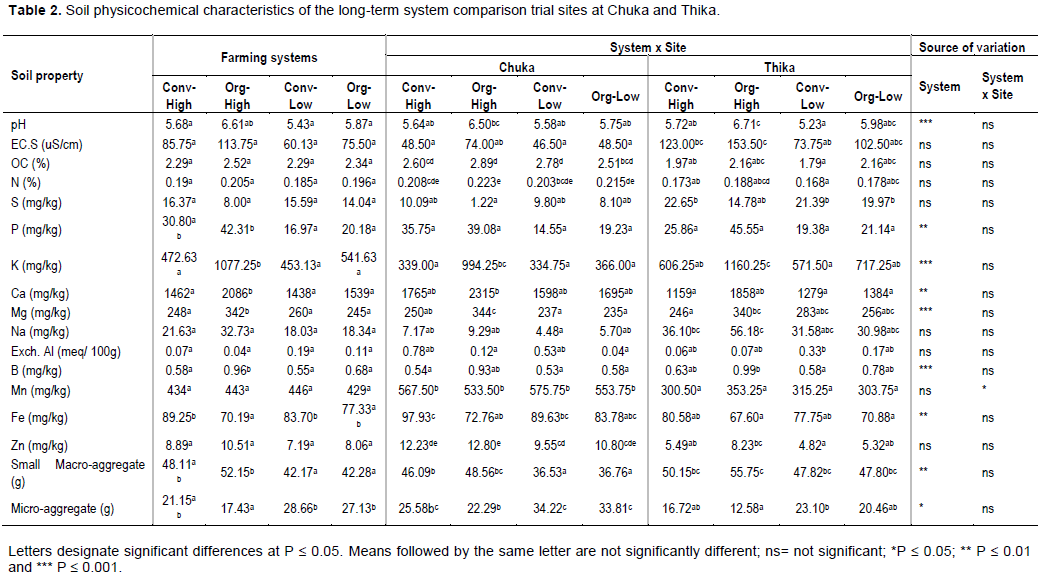

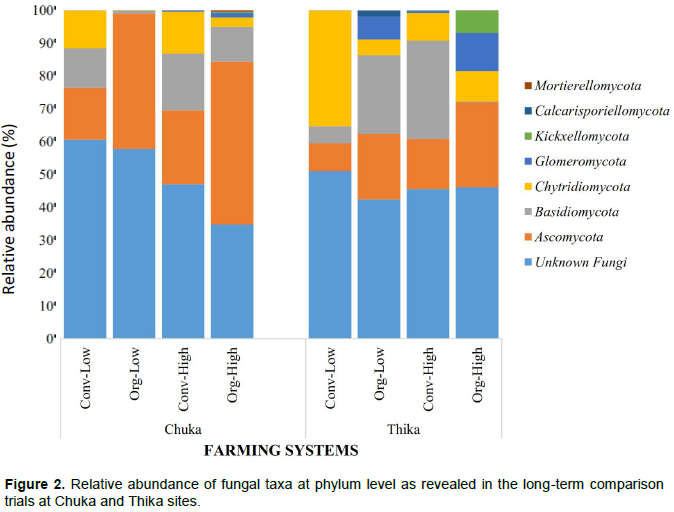


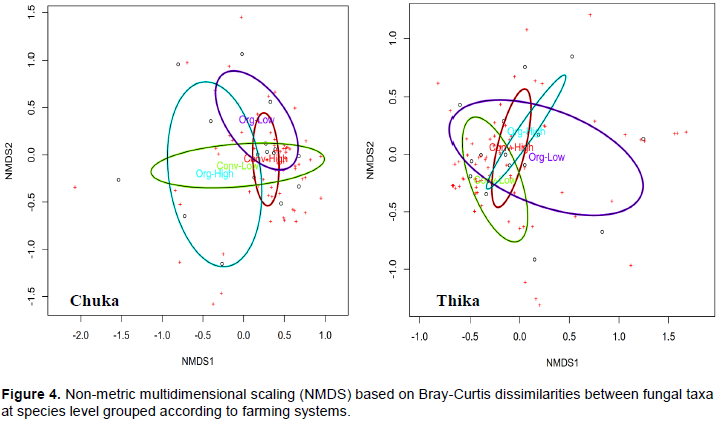

DISCUSSION
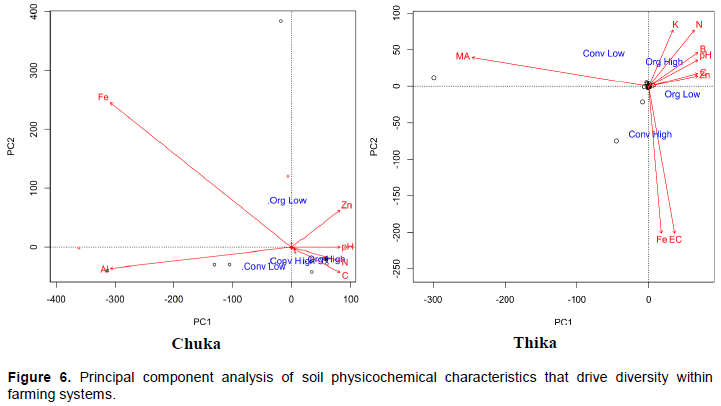
CONCLUSION
CONFLICT OF INTERESTS
The authors have not declared any conflict of interests.
ACKNOWLEDGEMENTS
ABBREVIATIONS
REFERENCES
|
Adamtey N, Musyoka MW, Zundel C, Cobo JG, Karanja E, Fiaboe KKM, Muriuki A, Mucheru-Muna M, Vanlauwe B, Berset E, Messmer MM, Gattinger A, Bhullar GC, Fliessbach A, Mäder P, Niggli U, Foster D (2016). Productivity, profitability and partial nutrient balance in maize-based conventional and organic farming systems in Kenya. Agriculture, Ecosystems and Environment 235:61-79. |
|
|
Anderson JM, Ingram JSI (1993). Tropical Soil Biology and Fertility: a Handbook of Methods. CAB International, Wallingford, UK. |
|
|
Bååth E, Anderson TH (2003). Comparison of soil fungal/bacterial ratios in a pH gradient using physiological and PLFA-based techniques. Soil Biology and Biochemistry 35:955-963. |
|
|
Balint M, Bahram M, Eren AM, Faust K, Fuhrman JA, Lindahl B, O'Hara RB, Opik M, Sogin ML, Unterseher M, Tedersoo L (2016). Millions of reads, thousands of taxa: Microbial community structure and associations analyzed via marker genes. FEMS Microbiology Reviews 40:686-700. |
|
|
Barea JM, Pozo MJ, Azcón R, Azcón-Aguilar C (2005). Microbial co-operation in the rhizosphere. Journal of Experimental Botany 56(417):1761-778. |
|
|
Bates D, Maechler M, Bolker B (2013). lme4: Linear mixed-effects models using S4 classes. R package version 0.999999-2. |
|
|
Berthrong ST, Buckley DH, Drinkwater LE (2013). Agricultural management and labile carbon additions affect soil microbial community structure and interact with carbon and nitrogen cycling. Microbial Ecology 66:158-170. |
|
|
Bloem J, Bolhuis PR, Veninga MR, Wieringa J (1995). Microscopic methods for counting bacteria and fungi in soil. In: Methods in Applied Soil Microbiology and Biochemistry. Alef K, Nannipieri P (Eds), pp. 162-173. Academic Press, London. |
|
|
Bolyen E, Rideout JR, Dillon MR, Bokulich NA (2018). QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Peer Journal Preprints 6:e27295v1. |
|
|
Callahan BJ, McMurdie P, Rosen MJ, Han AW (2016). Dada2: High-resolution sample inference from illumina amplicon data. Nature Methods 13(7): 581. |
|
|
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushma FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7:335-336. |
|
|
Dighton J (2003). Fungi in Ecosystem Processes. Marcel Dekker, New York. |
|
|
Dixon J, Gulliver A, Gibbon D (2001). Farming systems and poverty: Improving farmers' livelihoods in a changing world. FAO, Rome. |
|
|
Fierer N (2017). Embracing the unknown: Disentangling the complexities of the soil microbiome. Nature Reviews Microbiology 15:579-590. |
|
|
Fraç M, Hannula SE, BeÅ‚ka M, JÄ™dryczka M (2018). Fungal Biodiversity and Their Role in Soil Health. Frontiers in Microbiology 9:707. |
|
|
Gadd GM (2007). Geomycology: Biogeochemical transformations of rocks, minerals, metals and radionuclides by fungi, bio-weathering and bioremediation. Mycological Research 111(1):3-49. |
|
|
Gupta PK (1999). Soil, Plant, Water and Fertilizer Analysis. Agro Botanica Publishers, Bikaner, India, pp. 138-140. |
|
|
Harman GE, Howell CR, Viterbo A, Chet, Lorito M (2004). Trichoderma species-Opportunistic, a virulent plant symbionts. Nature Reviews Microbiology 2:43-56. |
|
|
Hartmann M, Frey B, Mayer J, Mäder P, Widmer F (2015). Distinct soil microbial diversity under long-term organic and conventional farming. International Society for Microbial Ecology 9:1177-1194. |
|
|
Ihrmark K, Bo€deker ITM, Cruz-Martinez, Friberg H, Kubartova A, Schenck J, Lindahl BD (2012). New primers to amplify the fungal ITS2 region evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiology Ecology 82:666-677. |
|
|
IUSS Working Group WRB (2006). World reference base for soil resources 2006 (2nd Edition). World Soil Resources Reports No. 103. FAO, Rome. |
|
|
Jaetzold R, Schmidt H, Hornetz B, Shisanya CA (2006a). Nairobi Farm Management Handbook of Kenya: Natural Conditions and Farm Management Information. Vol. II/C, East Kenya. Ministry of Agriculture, Nairobi, Kenya. |
|
|
Jaetzold R, Schmidt H, Hornetz B, Shisanya CA (2006b). Nairobi Farm Management Handbook of Kenya: Natural Conditions and Farm Management Information. Vol. II/B, Central Kenya. Ministry of Agriculture, Nairobi, Kenya. |
|
|
Johansson JF, Paul LR, Roger D (2004). Microbial interactions in the mycorrhizosphere and their significance for sustainable agriculture. FEMS Microbiology Ecology 48(1):1-13. |
|
|
Kazeeroni EA, Al-Sadi AM (2016). 454-Pyrosequencing reveals variable fungal diversity across farming systems. Frontiers in Plant Science 7:314. |
|
|
Kennedy JA, Powell HKJ (1986). Colorimetric determination of aluminium (III) with chrome azurol S and the reactivity of hydrolyzed aluminium species. Analytica Chimica Acta 184:329-333. |
|
|
Kojalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AF, Tedersoo L, Vrålstad T, Ursing BM (2005). UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytologist 166:1063-1068. |
|
|
Koljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Dueñas M, Grebenc T, Griffith GW, Hartmann M, Kirk PM, Kohout P, Larsson E, Lindahl BD, Lücking R, Martín MP, Matheny PB, Nguyen NH, Niskanen T, Oja J, Peay KG, |
|
|
Peintner U, Peterson M, Põldmaa K, Saag L, Saar I, Schüßler A, Scott JA, Senés C, Smith ME, Suija A, Taylor DL, Telleria MT, Weiss M, Larsson KH (2013). Towards a uniï¬ed paradigm for sequence-based identiï¬cation of fungi. Molecular Ecology 22:5271-5277. |
|
|
Lahlali R, McGregor L, Song T, Gossen BD, Narisawa K, Peng G (2014). Heteroconium chaetospira induces resistance to clubroot via upregulation of host genes involved in jasmonic acid, ethylene, and auxin biosynthesis. PLoS ONE 9(4):e94144. |
|
|
Lahlali R, Hijri M (2010). Screening, identification and evaluation of potential biocontrol fungal endophytes against Rhizoctonia solani AG3 on potato plants. FEMS Microbiology Letters 311(2):152-159. |
|
|
Lentendu G, Wubet T, Chatzinotas A, Wilhelm C, Buscot F, Schlegel M (2014). Effects of long-term differential fertilization on eukaryotic microbial communities in an arable soil: A multiple barcoding approach. Molecular Ecology 23(13):3341-3355. |
|
|
Lindahl BD, Ihrmark K, Boberg J, Trumbore SE, Högberg P, Stenlid J, Finlay RD (2007). Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytologist 173(3): 611-620. |
|
|
Lopes AR, Manaia CM, Nunes OC (2014). Bacterial community variations in an alfalfa-rice rotation system revealed by 16S rRNA gene 454-pyrosequencing. FEMS Microbiology Ecology 87(3):650-63. |
|
|
Madi L, Katan T, Katan J, Henis J (1997). Biological control of Sclerotium rolfsii and Verticillium dahliae by Talaromyces flavus is mediated by different mechanisms. Journal of phytopathology 87(10):1054-1060. |
|
|
McLaughlin DJ, Spatafora JW (2014). The Mycota. Systematics and Evolution Part A. Springer, Heidelberg. |
|
|
Mehlich A (1984). Mehlich-3 soil test extractant: A modification of Mehlich-2 extractant. Communications in Soil Science and Plant Analysis 15(12):1409-1416. |
|
|
Milner K (2014). Effects of Organic and Conventional Agricultural Practices on Soil Microbial Communities and Molecular Detection of Soil Borne Disease. Electronic Theses and Dissertations. 1213. |
|
|
Muriuki AW, Qureshi JN (2001). Fertilizer use manual. Kenya Agricultural Research Centre, Nairobi. |
|
|
Musyoka MW, Adamtey N, Muriuki AW, Cadisch G (2017). Effects of organic and conventional farming systems on nitrogen uptake and use efficiencies of potato, maize and vegetables in the sub humid region of Central highlands of Kenya. European Journal of Agronomy 86:24-33. |
|
|
Okalebo JR, Gathua KW, Woomer PL (2002). Laboratory methods of soil and plant analysis: A working manual. TSBF: Nairobi, Kenya. |
|
|
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens HH, Szoecs E, Wagner H (2012). Vegan: Community Ecology Package, R Package version 2.0-3. |
|
|
Persˇoh D (2015). Plant-associated fungal communities in the light of meta'omics. Fungal Diversity 75:1-25. |
|
|
Piepho HP (2004). An algorithm for a letter-based representation of all pairwise comparisons. Journal of Computational and Graphical Statistics 13:456-466. |
|
|
Purahong W, Wubet T, Lentendu G, Schloter M, Pecyna MJ, Kapturska D, Hofrichter M, Krüger D, Buscot F (2016). Life in leaf litter: Novel insights into community dynamics of bacteria and fungi during litter decomposition. Molecular Ecology 25:4059-4074. |
|
|
R Development Core Team (2012). A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. |
|
|
R Development Core Team (2014). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. |
|
|
Rillig CM, Muller LAH, Lehmann A (2017). Soil aggregates as massively concurrent evolutionary incubators. International Society for Microbial Ecology 11:1943-1948. |
|
|
Sambrook KJ, Fritsch EF, Maniatis T (1989). Molecular Cloning: A Laboratory Manual. (2nd Eds.). New York, USA: Cold Spring Harbor Laboratory. |
|
|
Schreiner RP, Bethlenfalvay GJ (1997). Mycorrhizae, biocides, and biocontrol 3. Effects of three different fungicides on developmental stages of three AM fungi. Biology and Fertility of Soils 24:18-26. |
|
|
Sharma-Poudyal D, Schlatter D, Yin C, Hulbert S, Paulitz T (2017). Long-term no-till: A major driver of fungal communities in dryland wheat cropping systems. PLoS ONE 12(9):e0184611. |
|
|
Singh M, Sarkar B, Biswas B, Bolan NS (2017b). Relationship between soil clay mineralogy and carbon protection capacity as influenced by temperature and moisture. Soil Biology and Biochemistry 109:95-106. |
|
|
Singh M, Sarkar B, Biswas B, Churchman J (2016). Adsorption-desorption behavior of dissolved organic carbon by soil clay fractions of varying mineralogy. Geoderma 280: 47-56. |
|
|
Six J, Elliott ET, Paustian K, Doran J (1998). Aggregation and Soil Organic Matter Accumulation in Cultivated and Native Grassland Soils. Soil Science Society of America Journal 62:1367-1377. |
|
|
Sommermann L, Geistlinger J, Wibberg D, Deubel A, Zwanzig J, Babin D, Schlüter A, Schellenberg I (2018). Fungal community profiles in agricultural soils of a long-term field trial under different tillage, fertilization and crop rotation conditions analyzed by high throughput ITS-amplicon sequencing. PLoS One 13(4):e0195345. |
|
|
Tedersoo L, Sa'nchez-Ram'ırez S, Ko˜ljalg U, Bahram M, Do ring M, Schigel D, May T, Ryberg M, Abarenkov K (2018). High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Diversity 90:135-159. |
|
|
Tedersoo L, Nilsson RH (2016). Molecular identification of fungi. In: Martin F (ed) Molecular mycorrhizal symbiosis. Wiley, Hobo-ken, pp 301-322. |
|
|
Tkacz A, Cheema J, Chandra G, Grant A, Poole PS (2015). Stability and succession of the rhizosphere microbiota depends upon plant type and soil composition. International Society for Microbial Ecology 9(11):2349-59. |
|
|
Wall DH, Bardgett RD, Behan-Pelletier V, Herrick JE, Jones TH, Ritz K, Six J, Strong DR, van der Putten WH (2012). Soil ecology and ecosystem services. 1st ed. Oxford University Press: USA. |
|
|
Wang H, Hyde KD, Soytong K, Lin F (2008). Fungal diversity on fallen leaves of Ficus in northern Thailand. Journal of Zhejiang University Science 9:835-841. |
|
|
Wang R, Zhang H, Sun L, Qi G, Chen S, Zhao X (2017). Microbial community composition is related to soil biological and chemical properties and bacterial wilt outbreak. Scientific Reports 7:343. |
|
|
White TJ, Bruns T, Lee S, Taylor JW (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: A Guide To Methods And Applications 18:315-322. |
|
|
Yu K, Zhang T (2012). Metagenomic and Metatranscriptomic Analysis of Microbial Community Structure and Gene Expression of Activated Sludge. PLoS ONE 7(5):e38183. |
|
Copyright © 2024 Author(s) retain the copyright of this article.
This article is published under the terms of the Creative Commons Attribution License 4.0


