Administration of drugs via oral route is the most common and convenient. Three major factors can affect drug absorption from this route: physiologic, physicochemical and formulation. Drug absorption mainly takes place in small intestine after oral administration. Within the small intestine, there are 2 major families of proteins involved in transport that have significant roles on drugs absorption: Solute-carrier and adenosine triphosphate-binding cassette (ABC) superfamilies. There are 48 ABC transporters in humans as well as seven subfamilies: ABCA to ABCG. ABCB1, ABCC1/2, and ABCG2 are widely described for their impacts in the absorption of drugs. Polymorphism on ABC transporters can significantly affect the absorption of drugs from the gastro intestinal (GI) tract. Some polymorphisms can result in over expression of the transporter in the small intestine and hence reduce absorption of drugs and some result in reduction in expression and increase in absorption. These polymorphisms are an important cause of adverse drug reactions and therapeutic failures.
Administering drugs utilizing the oral route is strongly preferred because of its convenience, and relative reduced cost (Engman, 2003). The small intestine is a major area of absorption for most orally administered drugs and so is the end for pharmaco-therapeutic approaches to regulate the oral absorption of drugs and the pharmacokinetics and pharmaco-dynamics parameters (Nakamura et al., 2008). The main mechanisms by which absorption occurs include transcellular or intracellular transport, paracellular or intercellular transport, active transport and vesicular transport or endocytosis (Salama et al., 2006).
Drugs which are administered orally must cross via the intestinal mucosa prior to arriving at the capillaries that drain to the portal vein causing first pass effect. The barrier of mucosa comprises polarized enterocytes that are intimately connected by way of firm junctions. Many drug transporters are detected in thecells (Estudante et al., 2013). Drug transporters are proteins that hold xenobiotics or endogenous compounds across membranes. They are divided as either uptake proteins or efflux, based on the way of transport. The two superfamilies of proteins involved in transport and that have significant roles on major pharmacokinetic profiles are the SLC and adenosine triphosphate-binding cassette (ABC) superfamilies (Regmi and Bharati, 2012).
This literature review focused on the ABC family members of transporters which are from the most widely investigated transporters involved in drug absorption and elimination and responses (Evans and McLeod, 2003). This superfamily members exploit adenosine triphosphate (ATP) as a source of energy, permitting to move molecules against a concentration grade. Drugs can be all together substrates and/or inhibitors of more than one efflux transporter, signifying that ABC transporters apply a dual role in detoxification at the intestine (Linton, 2007).
Genetic polymorphism (variation) is a difference in deoxyribonucleic acid (DNA) sequence among individuals, groups, or populations that contribute for a difference in drug absorption probably by affecting the transporter expression (Meletiadis et al., 2006). So many genetic polymorphisms in the transporters are investigated; some of them fascinated considerations as genetic factors linked with expression level and role in small intestine (Nakamura et al., 2008). The two important sites influencing the extent of drug reaching the systemic circulation followed by an administration are the intestine and/or liver, so transporter expression in these sites proposes that factors dictating their role will be important factors of oral drug pharmacokinetics. Factors influencing protein levels, genetic polymorphisms leading to improved or decreased purpose and co-administration with inhibitors are all significant opportunities where a transporter's capacity to transport molecules is changed (Estudante et al., 2013).
Introduction to ABC transporters
Structure
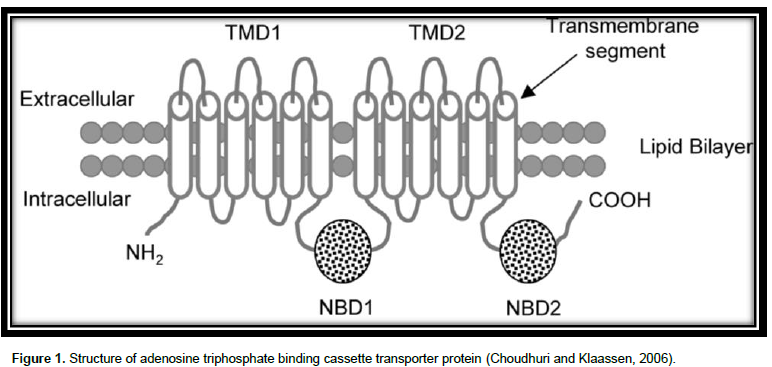
The superfamily of ABC proteins (Figure 1) are among the major protein families (Sharom, 2008). There are 48 ABC transporters in humans (Linton, 2007) which are categorized into 7 subfamilies: ABCA to ABCG according to the series homology. Among these, MDR1 drug-transporting P-glycoprotein (P-gp; ABCB1), multidrug resistance-associated 91 protein (MRP2; ABCC2), and breast cancer resistance protein (BCRP; ABCG2) have been well studied for the roles in drug disposition as well as response (Li and Bluth, 2011). The fundamental unit of an ABC transporter comprises 4 interior domains (Higgins, 2001), organized as either full transporters containing two transmembrane domain (TMD) and two nucleotide binding domain (NBD) or as half transporters having one of each domain. The half transporters come together as either homodimers or heterodimers to produce a useful transporter (Dean et al., 2001). The TMD, entrenched in the bilayer of membrane, differentiates a variety of substrates and goes through conformational modification to transport the substrate transversely across membrane (Yang, 2013) and provide specificity, whereas the NBDs attached as well undertake hydrolysis of ATP to force the movement of attached ligand (Linton, 2007).
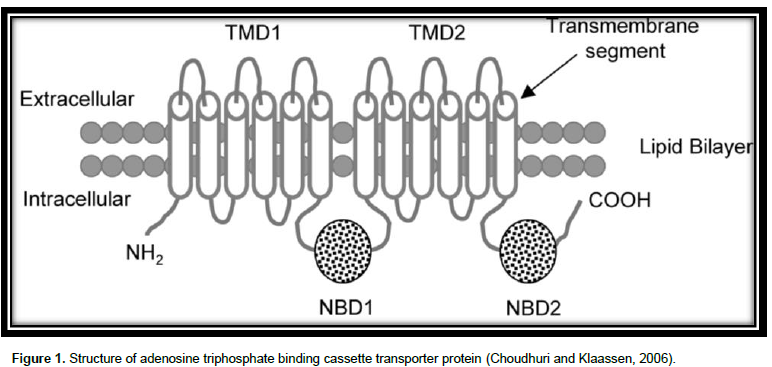
The TMD possess numerous alpha helices, which cross the lipid bilayer. Typically, there are six predicted membrane- spanning α-helices per domain (Higgins, 2001). The figure of alpha helices in a TMD varies based on the family. The attaching sites of ATP are situated on the membrane cytoplasmic (Cox, 2010).
The ABCB1 gene, the primary ABC transporter recognized, maps to chromosome 7q21.1 and comprise 28 translated exons and 27 introns. Previously called MDR1 or PGY1, ABCB1 was the initial human ABC transporter gene cloned and distinguished across its capacity to present a multi-drug resistant (MDR) phenotype to cancer cells which caused resistance to some therapy (Franke et al., 2010). The human MDR1 gene encodes a protein of 1280 amino acids which comprises two highly homologous halves. The molecular weight of P-gp is 170e180 kDa. This molecule holds 12 TMD and two putative ATP binding positions (Dallas et al., 2006).
MRP or ABCC family is the other member of ABC transporter and MRP2 is the main transporter of MRP family. MRP2 encodes a 190e200 kDapolytopic transmembrane protein consisting of 1545 amino acids and links to subfamily C of ABC transporter (Dallas et al., 2006). Twelve full transporters have been recognized so far. ABCC subfamily in human comprises ABCC1 through ABCC12, and nine of them belong to MRP transporters (Yang, 2013).
The ABCG2 (called BCRP, ABCP, or MXR) protein is an ABC half-transporter (Li and Bluth, 2011) which is composed of one transmembrane and one nucleotide linking fold section, called an NBF-TM. It is composed of 16 exons and 15 introns and is situated on chromosome 4q22 (Franke et al., 2010).
Expression and function
MDR1 drug-transporting P-glycoprotein: MDR1 is among the common efflux transporters uttered in MDR cancer cells and in numerous organs like intestine, liver, kidney and BBB. In the intestine of humans, P-gp is articulated in enterocytes apical membrane and the mRNA intensity is maximum in the jejunum, then in ileum and colon (Yang, 2013). It has a significant function by eliminating toxic materials/metabolites from cells. For example, the protein is extremely manifested in cells comprising BBB and most likely has a part in transport of toxic materials out of the brain, effectively stopping uptake (Franke et al., 2010). In the intestine, P-gp extrudes various drugs into the lumen, minimizing the rate as well as extent of absorption (Sharom, 2008). Poorly hydrophilic drugs with a polyaromatic skeleton and a positive or neutral charge are often the substrates for P-gp (Cox, 2010).
Multidrug resistance-associated protein: The protein is the most investigated ABCC family member (Cox, 2010) contained in the polarized cells apical membrane from range of human and rat tissues together with enterocytes of the small intestine, hepatocytes and renal proximal tubules (Dallas et al., 2006) where it can mediate the efflux of glucuronides, bilirubin and other organic anions, playing a function in the detoxification for various xenobiotic and endogenous compounds (Glavinas et al., 2004). The substrates for ABCC are several organic anions, especially conjugated compounds. In addition to conjugates, MRP2 moves amphipathic unionized compounds. Different investigations indicate that MRP2 mediated transport of unionized/positively charged substrates is motivated by the existence of low glutathione (Dietrich et al., 2003).
BCRP: ABCG2 was initially revealed, as indicated by the name, in cells of breast cancer. It was also named mitoxantrone resistance protein (MXR) due to one of its substrates (Dietrich et al., 2003). ABCG2 is usually manifested in the hepatocytes canalicular membrane, in the small intestinal, colon, placenta, lung, kidney, adrenal and sweat glands epithelia, as well as in the central nervous system (CNS) endothelia vasculature. It is very important for detoxification of host as well as defense against poisonous xenobiotics (Li and Bluth, 2011).
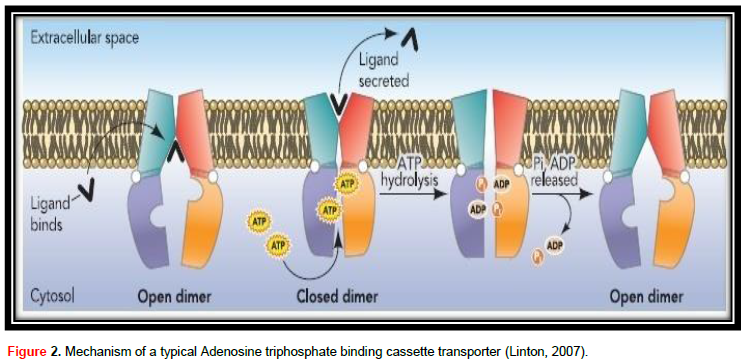
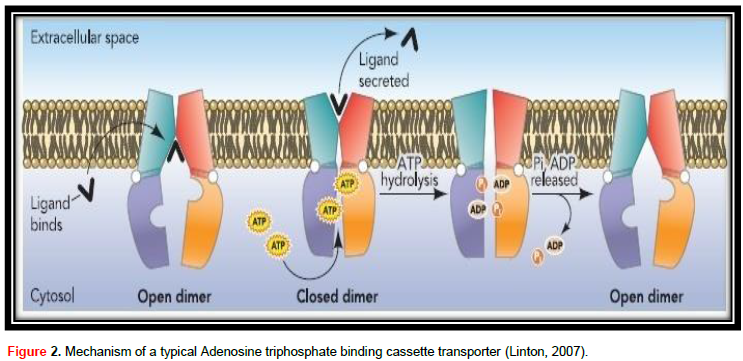
ABC transporter protein family, in spite of their great useful diversity, at their center they all contribute to the same field design and are thought to share a basically related to alternating access transport mechanism (Procko et al., 2009). The mechanism of a typical adenosine triphosphate binding cassette transporter is shown in Figure 2. The transport cycle is induced by the contact of substrate with the TMDs from the membrane intracellular face. The number of substrate-linking positions on TMDs is vague, though it is likely to be 2 (Higgins, 2001). Ligand attaches to the TMDs in the high-affinity open NBD dimer modification, stimulating higher affinity for ATP (Linton, 2007). Without nucleotide, NBDs are not together or open, but when ATP attaches to both NBDs they approach to form a tight dimer with two ATPase active locations at the interface (Procko et al., 2009), which on the other hand stimulates a high conformational alteration in TMDs adequate to move ligand. ATP hydrolysis induces dissolution of the stopped NBD dimer (Linton, 2007) and the transporter returns to the beginning of conformation (Procko et al., 2009).
Polymorphism of ABC drug transporters and influence on drug absorption from GI tract
MDR1 drug-transporting P-gp
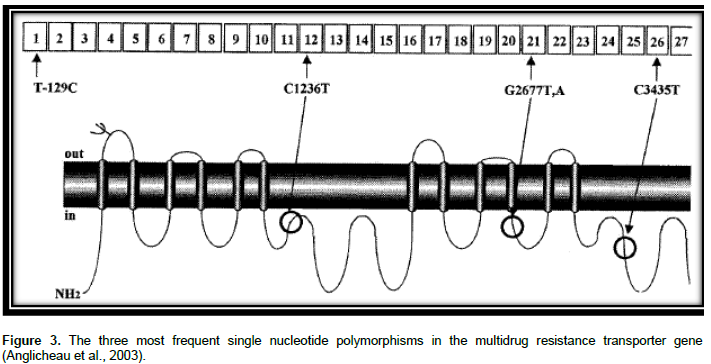
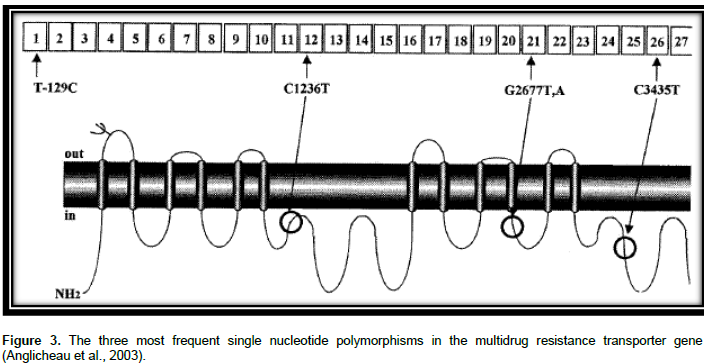
P-gp is substrate for many drugs together with bilirubin, several anticancer drugs, cardiac glycosides, immunosuppressive agents, glucocorticoids, and HIV-1 PIs (Evans and McLeod, 2003). ABCB1 genetic polymorphisms were indicated to alter the appearance of mRNA, protein expression and use of P-gp and change in substrate specificity (Sharom, 2008). Greater than 50 SNPs were recognized in the coding section of ABCB1 (Li and Bluth, 2011). C1236T in exon 12, G2677T/A in exon 21 and C3435T in exon 26 are three major SNPs in the ABCB1 gene as shown in Figure 3. The SNPs might influence the pharmacodynamics and pharmaco-kinetics of drugs that are P-gp substrates (Estudante et al., 2013).
C3435T: A SNP in exon 26 position 3435 (C3435T) was linked with the P-gp expression and function (Ekhart et al., 2009). Variation in the sequence of nucleotide from C to T at site 3435 did not cause an alteration of amino acid; however it is a quiet mutation situated in the wobble site of the codon. Although there is no alteration in induced protein, together the intensity of its function and expression may be changeable. For example, a double fall of intestinal P-gp had been seen in individuals who were homozygous for 3435T (Cox, 2010).
Effect on digoxin absorption
Digoxin is not affected by alteration of metabolism; it serves as a model substrate for phenotype-genotype interactions of MDR1 polymorphs. The digoxin uptake, a known P-gp substrate, is affected by intestinal P-gp induction. Consequently, digoxin plasma levels have been established to be significantly elevated in volunteers with a C3435 T nucleotide exchange in exon 26 (C3435T) of the MDR1 gene (Brinkmann and Eichelbaum, 2001). A sizable Dutch study (195 elderly patients) concerning chronic dosing of digoxin rather than single dose kinetics investigated the influence of MDR1 genotype on digoxin concentrations. The 3435C>T subjects were related with concentration of serum digoxin of 0.18 to 0.21 mcg/L per supplementary T allele. The findings of this study are in consistence with other study in healthy Japanese individuals. Individuals porting a T allele at 3435 had considerably lesser area under the curve than those homozygous for C at this site (Cox, 2010). Other study conducted on 32 healthy persons also found that the synonymous polymorphism in point 3435 affects the absorption kinetics of oral digoxin, the homozygous wild-type (CC) genotype eliminating digoxin more efficiently than the individuals by TT or CT alleles (Li and Bluth, 2011).
Effect on absorption of anticancer drugs
The substrates of P-gp comprises anticancer drugs like vincristine, doxorubicin, vinblastine, etoposide, daunorubicin, and paclitaxel (Novoa et al., 2005). Reports on individual SNPs at 3435 had significantly showed lower exposure to SN-38 (7-ethyl-10-hydroxycamptothecan), irinote can activate metabolite by carboxylesterase enzymes (Cox, 2010), signifying that the variant haplotype is linked by high efflux action (Franke et al., 2010).
Effect on absorption of antiretroviral agents
Most protease inhibitors of HIV-1 are substrates of the P-gp system, which affects their uptake as well as body distribution (Novoa et al., 2005). The MDR1 C3435T genotype influences the absorption constant of indinavir suggesting that P-gp might be concerned in its variability of pharmacokinetics profile. Result of study conducted by Solas et al. (2007) indicated that patients having the MDR1 C3435T genotype had an extensively elevated ka (two-fold increase) compared with patients having the C3435C genotype Solas et al. (2007). Studies conducted on other antiretroviral drug, nelfinavir, also show that T allele at position 3435 in exon 26 is linked with a superior level of P-gp purpose. Individuals with a homozygous T genotype at exon 26 had a lower level of nelfinavir in plasma in comparison to those individuals with the homozygous C genotype. This result suggested the T allele is linked by a superior concentration of P-gp purpose (Sankatsing et al., 2004).
Effect on other drugs
As a result of hydrophobic structure, antiepileptic drugs might be substrates for P-gp transporter (Buzoianu et al., 2009). Investigations of 108 patients for phenytoin concentrations of serum along with MDR1 C3435T gene polymorphism by Ponnala et al. (2012), indicated steady raise in phenytoin concentrations of serum from homozygous C allele, heterozygous CT, and homozygous T allele. Genotype and serum phenytoin concentration examination as indicated by the MDR1 C3435T gene polymorphism influences serum phenytoin concentrations. The mean serum phenytoin concentrations in seizure re-occurrence genotype groups were 8.65, 9.51, and 17.54 mg/ml for CC, CT, and TT genotypes, respectively (Sailaja et al., 2010).
G2677T/A: The non-synonymous G2677T/A SNP is located in exon 21, which represents an amino acid change of alanine by serine (G2677T) or threonine (G2677A) (Ala893Ser/ Thr) (Brambila-Tapia, 2013) in such a manner that the lipophilic residue (Ala) is altered to hydrophilic residue (Ser, Thr) presenting higher resistance to different drugs such as adriamycin and vinblastine (Sailaja et al., 2010). This SNP has shown controversial results in functional effects (Brambila-Tapia, 2013). A Report of study conducted by Lamba et al., 2006 indicated that those who were with the 2677 TT genotype had lesser P-gp messenger RNA expression than those who had 2677 GG genotype (Sailaja et al., 2010). A study conducted by Green et al. (2006) showed that the missense SNP, G2677T/A, correlated with the outcome of paclitaxel treatment. The study compared the wild-type and heterozygous (G/G and G/T) with the homozygously mutated (T/T and T/A) patients and their relation to treatment outcome. A statistically significant association was obtained between homozygously mutated patients as well as successful treatment with paclitaxel. The occurrence of the T and A alleles in the group of patients with a good response was also significantly upper than in poor responders (32 of 56 compared with 18 of 50). One explanation could be that the G2677T/A polymorphism have a functional consequence on P-gp mediated paclitaxel transport. The better response might be due to a lower efflux of paclitaxel from the tumor cells or an increased absorption, giving higher plasma concentrations (Green et al., 2006). On the contrary to the aforementioned study, G2677T SNP was linked with improved P- gp role in vitro and lesser plasma fexofenadine levels in humans (Evans and McLeod, 2003).
C1236T: The synonymous C1236T SNP is located on exon 12 which encodes for the TM6 region, which is essential for substrate binding (Brambila-Tapia, 2013). Variability in MDR1 on exon 12 was linked with variations in plasma drug concentrations as well as response to ART. Bellusci et al. identified genotypes in 100 blood donors and 38 HIV-1-infected patients. Every patient took highly active antiretroviral therapy (HAART) with LPV/r at the moment of lopinavir plasma concentration quantification, prior to and between 1 and 2 h following the next dose of drug administration. CD4 (+) T-cell counts and plasma viral load had been examined before and after the start of LPV/r. The result suggests that the MDR1 C1236T SNP drastically lowers LPV plasma con-centration, influencing the virological response to HAART. Heterozygotes C1236T could have a changed concentration of P-gp expression/activity in enterocytes and CD4 (+) T lymphocytes which confines the absorption of LPV, causing a weakened virological suppression (Bellusci et al., 2013).
Breast cancer resistance protein, BCRP (ABCG2)
Breast cancer resistance protein (BCRP) is one of the newer ABC transport proteins to be examined. The transporter was first identified in drug-resistant breast cancer cells. It actively forces out a broad range of chemically dissimilar hydrophobic compounds from the cells, as well as cytotoxic compounds like topotecan, mitoxantrone, SN-38, flavopiridol, and methotrexate (Li and Bluth, 2011), non-chemotherapy drugs like prazosin, dipyridamole, statins, glyburide, quercetin, temocapril, an ester-type prodrug of temocaprilat, sulfate conjugates, nitrofurantoin, porphyrins, some fluoroquinolones, antivirals such as lamivudine, zidovudine and efavirenz, rifampicin, ciprofloxacin, quercetin, sulfasalazine and nontherapeutic compounds such as dietary flavonoids (Estudante et al., 2013).
Over 80 SNPs, missense and frame shift mutations in the ABCG2 gene were recognized in dissimilar racial group (Sharom, 2008). Functional SNP in exon 5 of the ABCG2 gene, in which a C to A nucleotide transition at site 421 (ABCG2421C.A) exist, causes a nonsynonymous variant protein with a glutamine to lysine amino acid replacement in codon 141. The ABCG2C421A variant was linked with small ABCG2 expression levels and changed substrate specificity, and varied diflomotecan and topotecan pharmacokinetics (Li and Bluth, 2011).
Another study indicated that a higher ratio of patients existed more than 15 months with docetaxel-based therapy in the occurrence of the ABCG2 C421A polymorphism. The consequence of ABCG2 poly-morphisms on docetaxel disposition is unidentified. The enlarged survival seen in those with an ABCG2 C421A polymorphism can recommend a fewer functional drug efflux pumps, causing improved absorption and concentrations of intracellular docetaxel, as well as enhanced cytotoxic activity (Ekhart et al., 2009).
A study conducted by Zhang et al. (2006) also showed that the pharmacokinetic profiles of rosuvastatin revealed a appreciably high dissimilarity between the two genotyped groups. In the C421A along with A421A mutant group, the area under the curve (0 to 72) and the area under the curve (0 to ∞) of rosuvastatin had been about 80% elevated than that in the C421C wild-type, and the Cmax was about 90% better than that in the C421C wild- type.
The study conducted by Urquhart et al. (2007) shows the influence of general SNPs in BCRP to the disposition of sulfasalazine in healthy individuals. Sulfasalazine is a substrate of BCRP and utilized in the therapy of ulcerative colitis. In the study, individuals with 34GG/421CA genotype, sulfasalazine AUC0- N and Cmax values were considerably better than those obtained from wild-type 34GG/421CC individuals. Fascinatingly, the lone subject transporting variant alleles, 34A and 421A, demonstrated a 4.8-fold raise in the AUC and a 4.4-fold raise in Cmax in comparison to wild-type controls. The data of in vitro expression proposed lower cell surface expression of the 421C>A variant as a major influencing factor of lower BCRP mediated transport of sulfasalazine in individuals having an A allele at site 421.
A study conducted Zamboni et al. (2006) also showed that the ABCG2 421C>A genotype radically influenced 9-aminocamptothecin (9AC) pharmacokinetics. The 9-nitrocamptothecin (9NC) is an orally administered camptothecin analogue.
MDR-associated protein family
ABCC2 plays a major part in the transport of drugs like leukotriene, oxidized glutathione, vincristine, cisplatin, methotrexate, daunorubicin, etoposide, glutathione, glucuronide, and sulfate conjugates (Dallas et al., 2006). Most ABCC2 polymorphisms are quite rare in the general population, however, 24C>T, 1249G>A, and 3972C>T are all relatively common in healthy individuals. The functional importance of ABCC2 polymorphisms remains unclear. Few studies assessed results of polymorphisms in this gene and those that have failed to find any functionally significant effects (Sissung et al., 2012). The pharmacokinetics of irinotecan as well as 9- nitrocamptothecin and its metabolite 9-amino-camptothecin in cancer individuals were not considerably influenced by the polymorphism of ABCC2 –24C>T (Zamboni et al., 2006). ABCC2 gene mutations are linked with the rare autosomal recessive disorder Dubin-Johnson syndrome (DJS). The mutations might form DJS through a diversified system. The common understandable is creation of nonfunctional forms of the protein, that causes the incapability for hepatocytes to produce conjugated bilirubin into the bile (Choudhuri and Klaassen, 2006). Numerous mutations linked with DJS arise on the ATP linking section, which is significant for protein purpose. The intestinal expression and function of ABCC2 in DJS patients are lower. To explain its clinical importance in drug absorption, genotype-haplotype analysis of ABCC2 is necessary (Nakamura et al., 2008).
Application of ABC transporters polymorphism on clinical practice
The vital function by ABC multidrug efflux pumps in carrying tissues from exogenous toxins is broadly documented. These associated transporters determine the drugs uptake as well as release to their tissue target (Sharom, 2008). Polymorphism result in altered drug absorption and then altered drug response. The detection of polymorphisms elucidating separate drug phenotypes transporter proteins put into the acceptance of personal changes (personalization) of drug treatment and enhances drug safety and efficacy (Ma and Lu, 2011).
Personalized medicine is a medical model with molecular profiling technologies for following the correct pharmacologic approach for the appropriate individual at the appropriate time. Accurate prediction about drug response is crucial for individualized treatment, is best made by combining an individual’s genetic data with clinical findings and classifying individuals into subpopulations that differ in their response to an exact drug. A pharmacogenomics advancement may permit a precise drug therapy to be applied to genetically distinct subsets of patients and may direct to a new treatment categorization at the molecular stage (Eichelbaum et al., 2006).
The understanding about occurrence of functionally significant SNPs in the ABC gene in the population is a considerable idea for scheming prospect pharmacokinetic and pharmacodynamic investigations conducted with ABC substrates. Pharmacodynamic approaches will permit rising power of the investigations with lower economic expenses and ethical threat for the participating individuals at the right time. The practicing clinicians should be further conscious of the high frequency of functional polymorphisms, particularly when in view of treatment with drugs – substrates with a narrow
therapeutic window. Digoxin is one of the model drugs, where special carefulness is necessary in those with lower activity of transporters to avoid development of severe adverse drug reactions (Pechandova et al., 2006).
Transport activity of ABC is known to influence oral uptake of substrate drugs and polymorphism may sometimes result in treatment failure, for example on HIV, cancer and epilepsy. Identification of these polymorphisms allows improving treatment response through individualization of therapy (Sharom, 2008).
Future perspectives
Drug transporters amend the drugs absorption, distribution, and elimination by regulating the uptake and efflux of drugs in cells. Rising proof shows genetic polymorphisms of transporters can cause intense influence on drug absorption, efficacy, and safety. The vast of investigations had resulted from grim experimental restrictions, such as model variety, sample size, puzzling factors and genotype/phenotype errors, and a complete set of suggestions to circumvent such troubles in the prospect existed. Confirmatory effect of the ABC SNP is desirable. Large-scale genotype-phenotype association trials are required to raise our awareness of the effects of SNP on clinical responses.
A recent review about pharmacogenomics highlights the need for depth analysis of the available whole genome association study data, more advances in technologies and sequencing the complete genome (whole-genome sequencing) to identify low frequency or rare variants that are associated with HIV infection. In addition, data collections should be extended to as many populations as possible to enhance diversity. It is particularly important in African populations due to the high HIV prevalence rate. In the future, the individualized medicine will possibly comprise a mutual approach by means of the knowledge of drug, virus and host factors information to guide the personalized prescription in which the right drug will be given to the right person.
The function of polymorphism in response to drug treatment and disease vulnerability is an increasing area that will visibly be significant in the future as the eventual target of individualized drug is followed.