ABSTRACT
Conventional pharmacokinetic profiles of drugs are described mainly by plotting drug plasma concentration versus time. However, topical drugs are designed to target local tissue and, as such, have limited systemic absorption. For these drugs, the pharmacokinetic measurements in the local tissue (skin) are of more importance than systemic measurements. In this context, this study aims to develop and discuss the appropriate bioanalytical methodology by high performance liquid chromatography (HPLC) and application to ex vivo DPK metronidazole (MTZ) study based on the resolution - RE no. 899/2003 and Collegiate Board Resolution – RDC no. 27/2012 guidelines from the National Health Surveillance Agency of Brazil (ANVISA) and the FDA Guidance for Industry Bioanalytical Method Validation (2013). The average of recovery was 98.71% in a linear range from 0.1 to 20 μg/mL. The intraday and interday precision and accuracy were within specified limits to bioanalytical methods. MTZ was not detected in the receptor solution after 6 h, demonstrating that during this time, the drug is not able to cross the skin at a flow, resulting in a concentration higher than 0.1 μg/mL, which is the lower limit of quantification of this method. The amount of MTZ found in the pig SC was 20.91±7.02 µg/cm2 (n=14). Based on these results, it can be concluded that the validation of the drug dosage methodology described here was sensitive, precise and accurate and was successfully applied to analyze ex vivo DPK samples and is an important step for ensuring the reliability of these analyses.
Key words: Bioanalytical method, dermatopharmacokinetic (DPK), metronidazole, tape stripping.
The conventional pharmacokinetic profiles of drugs are described mainly by plotting drug plasma concentration versus time. However, some drugs, such as local anesthetics, antifungals, antibacterial agents, antiprotozoal agents and topical corticosteroids, are designed to target the local tissue and, as such, have limited systemic absorption. For these drugs, the pharmacokinetic measurements in the local tissue (skin) are of more importance than systemic measurements (Chik et al., 2010). Along this line of reasoning, the dermatopharmacokinetic (DPK) concept involves the determination of the amount of drug present in the stratum corneum (SC) as a function of time post-application and post-removal of formulation (Wiedersberg, 2006). The determination of drug concentration in the SC is made by the sequential removal of thin layers of SC with adhesive tape and later extraction and analysis (N’dri-Stempfer et al., 2003). This procedure can be performed in vivo or ex vivo using either skin obtained from surgery or animal skin, with pig being most commonly used. There are several reports on determining the drug concentration in the SC through tape analysis. However, the majority of the methods used were not described as validated according to an official guide. Some of the reports describe only recovery tests, selectivity, and linearity (Alberti et al., 2001; Pershing et al., 2002; Lodén et al., 2004; Benfeldt et al., 2007; Herkenne et al., 2006; Aggarwal et al., 2013; Herkenne et al., 2007; Pershing et al., 1993; Wana et al., 2015; Nagelreiter et al., 2015; Dai et al., 2015; Patel et al., 2009; Hathout et al., 2011). In addition to the analytical method validation approach, other parameters, as bioanalytical method evaluation, should be well evaluated during method validation, such as the recovery, precision and accuracy of the whole analytical range of the calibration curve (low, medium and high quality control), considering mainly that the quantity of analyte measured in each tape, obtained from the permeation process in SC, can highly range as a kinetic process. The parameters in their entirety should also be observed through a bioanalytical optic because the drugs are extracted from a biological matrix with a high possibility of interference from endogenous compounds, especially in HPLC with ultraviolet (UV) detection. Considering that this DPK method is recognized as a promising approach to evaluating the bioequivalence of topical drug products, this study aims to develop and discuss the appropriate bioanalytical methodology by HPLC-UV for application of ex vivo DPK metronidazole (MTZ) study based on the RE no. 899/2003 and RDC no. 27/2012 guidelines from the National Health Surveillance Agency of Brazil (ANVISA) and the FDA Guidance for Industry Bioanalytical Method Validation (2013).
Chemicals and reagents
The active pharmaceutical ingredient (MTZ) was obtained from Hubei Hongyuan Pharmaceutical/Hong Kong/China, sodium phosphate monobasic was purchased from Vetec/ Rio de Janeiro/Brazil; acetonitrile was obtained from J.T. Baker/México, and standard MTZ was supplied by the National Institute for Quality Control in Health–Oswaldo Cruz Foundation (INCQS - FIOCRUZ), lot: W2F01.
Formulation
The tested formulation was Rozex® (reference topical drug formulation 0.75%) (Brasil, 2015).
Standard solutions
Stock solution was prepared for method validation at a target concentration of 100 µg/mL MTZ free base in acetonitrile : water (50:50, v/v) to analyze ex vivo DPK data (samples from SC layers extractions of pig skin). A series of working solutions of MTZ were prepared to obtain standard solutions with concentrations of 0.1, 0.5, 1, 5, 10, 15 and 20 µg/mL and quality controls (0.1, 0.3, 5, 16 and 50 µg/mL) for validation procedures and sample analysis.
Chromatographic conditions
The HPLC system employed was a Shimadzu High Performance Liquid Chromatograph equipped with two isocratic pumps (LC20AD), column oven (CTO20AD), autosampler (SIL20AC) and diode array detector (SPDM20AD) set at 320 nm for quantification and at a range of 200 to 400 nm to UV screening. Separation was performed on a Shimadzu© 150 x 4.60 mm C18 (5μm) reversed-phase column and a 4 x 4 mm C18 guard column at 35°C. A monobasic sodium phosphate buffer 20 mM pH 3.0 : acetonitrile (88:12, v/v) was used as the mobile phase at a flow rate of 1 mL/min and a sample injection volume of 20 μL.
Sample preparation
After removal of layers of the stratum corneum from pig skin by tape stripping, the tapes were transferred to glass tubes and 1.0 mL of acetonitrile was added, followed by shaking for six h at 32°C. Next, the samples were subjected to ultrasound (Quimis©) for 30 min. They were subsequently centrifuged (Centribio©) at 75.6 RCF for 30 min. They were then filtered using 0.45 μm Millex© HV-PVDF filters and analyzed using the developed HPLC method. The use of internal standard was not necessary according sample preparation procedure (dilution of matrix) and detection by UV at 320 nm.
Validation of the analytical method
The HPLC method was validated following the Resolution RE no. 899/2003 for analytical methods and Resolution RDC no. 27/2012 for bioanalytical methods (ANVISA Brazil, 2003), and the FDA Guidance for Industry Bioanalytical Method Validation (2013). The authors evaluated the selectivity based on the Resolution RE no. 899/2003; and linearity, selectivity, precision and accuracy based on the Resolution RDC no. 27/2012 and the FDA Guidance for Industry Bioanalytical Method Validation (2013).
Linearity
To measure the linearity, three different calibration curves were prepared. Each calibration curve was obtained at seven concentrations of MTZ (0.1 – 20.0 μg/mL). The lower limit of quantification (LLOQ) and upper limit of quantification (ULOQ) were established to cover all intervals of the DPK profile. Calibration curves were fitted using least squares linear regression analysis. The back-calculated values were obtained and precision and accuracy were observed according to the specification of ±20% for LLOQ and ±15% for other levels.
Selectivity
The selectivity was assessed by three samples of blank tape (Scotch Booktape #845, 3M Co., St Paul, MN) and six different stratum corneum from pig skin.
The selectivity of the analytical method was also evaluated by monitoring MTZ in the presence of formulation components, MTZ gel (0.75%), and placebo gel. They were diluted in a theoretical concentration of 20 μg/mL in acetonitrile : water (50:50, v/v) and scanned using a photo diode array detector in a range of 200 to 400 nm following HPLC separation.
Precision and accuracy
Precision was determined both within a single analytical sequence (intra-assay) and among 3 separate analytical sequences (inter-assay). Each analytical sequence included 5 replicates in five concentrations: experimental LLOQ, low (LQC), medium (MQC), high (HQC) and dilution (DQC) quality controls at 0.1, 0.3, 5.0, 16 and 50μg/mL, respectively. The DQC was diluted from 50 to 16 μg/mL prior to sample preparation (Anvisa Brazil, 2012). To obtain the samples, tapes were contaminated with the respective concentrations in five replicates.
Recovery
Absolute recovery of MTZ from tapes and skin was evaluated by exposing blank with SC tape and skin slices without SC to known concentrations (0.3, 5.0 and 16.0 µg/mL), the MTZ solutions were applied to samples and dried at room temperature (21 to 23°C) for 6 h. Following complete evaporation, samples handling were proceed according to sample preparation.
Stability of samples
The stability of the samples stored in solutions or in tapes was evaluated during method validation. MTZ samples (5 μg/mL in each tape), prior to tape extraction, were analyzed after storage for periods of 24, 48 and 72h (short-term stability), and 7 and 15 days (long-term stability) at room temperature (21-23°C). Sample solutions (after extraction) were kept in the autosampler at 4°C and analyzed after a period of 24, 48 and 72 h. All samples were compared with nominal concentrations and freshly prepared MTZ samples at the same concentration level (0.3 and 16 µg/mL). The temperatures and test durations were chosen based on routine analysis of samples.
Method application (DPK in vitro)
Drug administration
Formulations were applied and distributed as evenly as possible with finger gloves, providing approximately 56.5 mg of product/cm2 of exposed skin area, corresponding to 423.75 µg/cm2 of MTZ from Rozex®.
Tape stripping procedure
The MTZ concentration profile across the SC following application was determined by sequential removal of this outer skin layer by tape stripping (Scotch Book Tape, 3M, St. Paul, MN). 30 strips were taken from each treated site on pig skin. Tapes 1 to 14 were extracted individually but tapes 15-17, 18-20, 21-24, 25-27 and 28-30 were grouped for extraction. All tapes were subsequently analyzed, no strips were discarded, and it was assumed that any drug not removed by the surface cleans process at the end of the treatment would eventually be bioavailable to the skin.
Analysis of DPK in vitro
Fresh pig ears were obtained from three different pigs at a local abattoir (Paulista, Pernambuco, Brazil); ears were removed post-sacrifice but before the carcass was exposed to the normal high-temperature cleaning procedure. Ears were washed with water and any visible hair was trimmed with scissors. Next, the skin was dermatomed at 750 µm (Zimmer air dermatome, Dover, Delaware). The membranes were stored at -20°C until used. The skin was allowed to thaw for 30 min prior to use. Excised pig ear skin was stretched across the aperture of a vertical Franz cell (Vision© Microette), held together in a clamp; 1.77 cm2 of skin was exposed. The dermal side of the skin was bathed in receptor fluid monobasic sodium phosphate buffer (20 mM), pH 7.4 and stirred by a magnetic bar. The formulations were applied to the donor compartment; after 6 h, the skin was cleaned and the tape stripping procedure was performed. Next, 1 mL of the receptor solution was collected and analyzed by HLPC.
Selection of receptor solution: The solubility of MTZ was determined by preparing dispersions containing excess drug (400 mg) in 5 mL of different solutions, such as phosphate buffer (20 mM) pH 7.4, water : ethanol, 50:50, v/v, phosphate buffer (20 mM) pH 7.4 with 0.5% polyoxyethylene 20 oleyl ether (Brij 98©), and phosphate buffer (20mM) pH 7.4 with 0.5% Tween 80©.These dispersions were homogenized by stirring at a controlled temperature (32 ± 2°C) over a period of 24 h and then centrifuged (75.6 RCF) for 30 min, after which an aliquot of supernatant was removed and filtered through a 0.45 µm membrane filter. The concentration of MTZ was determined by UV spectrophotometry (320 nm) using a solvent and a suitable calibration curve for each medium.
Validation
Linearity
Evaluation of the data curves using the least squares method showed that the method was linear in the range of 0.1 to 20 μg/mL. The equation obtained by a linear regression was y = 25366x - 2190 and the determination coefficient r² = 0.9998. Analysis of variance (ANOVA) showed that lack of adjustment was not observed, according to the F value (0.00029). The accuracy of the back-calculated results was between 92.61 and 106.64%. This method was reliable and valid. The LLOQ was 0.1 μg/mL. This method was demonstrated to be suitable for determining MTZ in DPK samples ex vivo, since MTZ could be analyzed in all the tapes.
To establish the ideal concentration range, the expected concentrations should be known. For the DPK description, as a kinetic process, the analytical method needs to provide a range to avoid sample dilution process and samples below LLOQ. The variation of concentrations of DPK samples collected should result between the LLOQ and ULOQ (Ema, 2009).
Selectivity
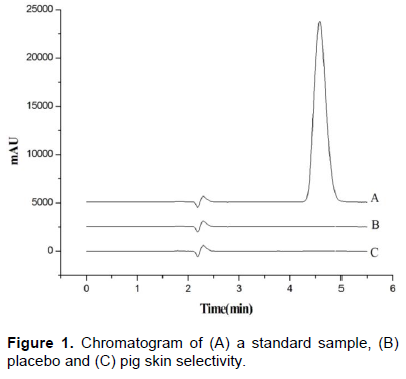
The method showed selectivity, and there was no analytical interference from other formulation components (placebo) or biological interference (layers of the stratum corneum and epidermis and dermis without SC from pig skin) as compared to standard samples. Figure 1 shows the chromatograms obtained for selectivity. In addition, an analysis with diode array detector showed no co-elution peaks at MTZ retention time, peak purity index was 1.000000 (single point threshold 0.999967).
Precision and accuracy
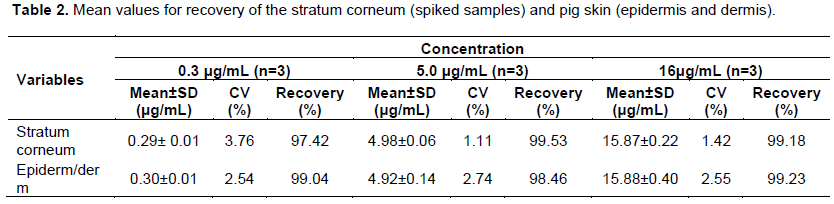
Intra- and inter-day precision and accuracy were satisfactory. The data obtained from pig tapes (Table 1) are within the acceptable limits set by the guidelines for validation of bioanalytical methods (FDA, 2002; Resolution No. 899, 2003).
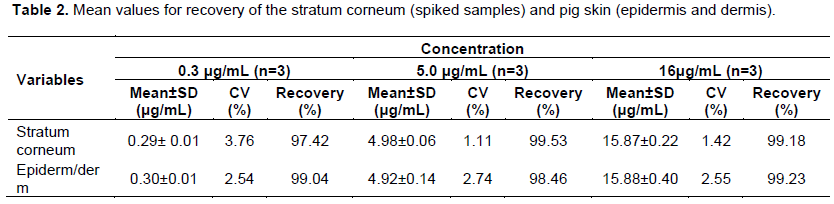
Recovery
Presents mean values for the recovery of MTZ from pig skin stratum corneum and pig skin (epidermis and dermis without SC) in three different concentrations Table 2. The results confirm that the extraction process was efficient for the recovery of MTZ in both pig skin layers, since the substance could be recovered at significant amounts, regardless of whether the concentration tested was low, medium or high. This test is highly important considering that the drug concentration in the deeper layers of SC is much lower than in the surface layers where the formulation is applied. It is noteworthy that the recovery study should be performed by applying a known solution on the skin and leaving it there for a standardized amount of time in order to begin the extraction procedure. As the drug pathway (intra or inter-cellular) following skin application is not known, the use of ultrasound during the recovery procedure is desirable, for the higher drug concentration is recovered from the skin.
Stability of samples
The stability data of samples in various forms of storage (MTZ+SC dried in tapes and MTZ+SC in extraction solution) demonstrated that the concentrations calculated for the controls were not significantly modified as no degradation higher than 3.0% was observed. Thus, the samples can be considered as stable for up to 15 days under the evaluated conditions. This evaluation is particularly important when the drug can easily degrade in the presence of biological matrix and during extraction using organic solvents (acetonitrile), high temperature (32°C), and ultrasonic bath.
DPK in vitro
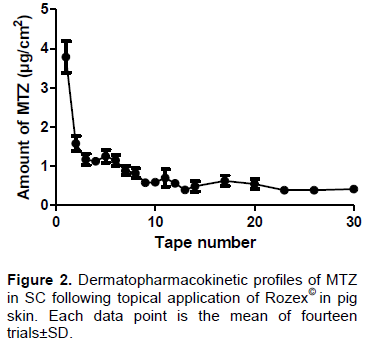
The MTZ solubility in tested solutions at 32±2°C was 8.90, 14.46, 7.13, 10.13, 8.91 and 11.08 mg/mL, respectively, for phosphate buffer (20 mM) pH 7.4, water alcohol solution (water : ethanol, 50:50 v/v), phosphate buffer (20 mM) pH 7.4 with 0.5% polyoxyethylene 20 oleyl ether (Brij98©), and phosphate buffer (20 mM) pH 7.4 with 0.5% Tween 80©. Considering that all the solutions can ensure the sink condition for in vitro DPK procedures, phosphate buffer (20 mM), pH 7.4 was selected as a receptor medium. MTZ was not detected in the receptor solution after 6 h, demonstrating that during this time, the drug is not able to cross the skin at a concentration higher than 0.1074 μg/mL, which is the quantification limit of this method. The MTZ amount found in the SC (n=14) was 20.91±7.02 µg/cm2 and the profile is shown in Figure 2.
Based on these results, it can be concluded that the validation of drug dosage methodology described here is sensitive enough to analyze ex vivo DPK (pig skin) samples, and is an important step for ensuring the reliability of these analyses. The inclusion of the stability test of the drug in a matrix such as tape or extraction solution is necessary, regardless of the physical and chemical characteristics of the drug and the recovery study should be performed with different drug concentrations to avoid non-corrected concentrations of unknown samples and demonstrate the necessity of use of internal standard to correct variations in the sample preparation. The linearity, precision and accuracy evaluations by the bioanalytical approach were also important for monitoring the quality of analysis of a kinetic process.
REFERENCES
|
Aggarwal N, Goindi S, Khurana R (2013). Formulation, characterization and evaluation of an optimized microemulsion formulation of griseofulvin for topical application. Colloids and Surfaces B. Biointerfaces 105:158-166.
Crossref
|
|
|
|
Alberti I, Kalia YN, Naik A, Bonny JD, Guy RH (2001). In vivo assessment of enhanced topical delivery of terbinafine to human stratum corneum. J. Controlled Release 71(3):319-327.
Crossref
|
|
|
|
|
Benfeldt E, Hansen SH, Vølund A, Menné T, Shah VP (2007). Bioequivalence of topical formulations in humans: evaluation by dermal microdialysis sampling and the dermatopharmacokinetic method. J. Invest. Dermatol. 127(1):170-8.
Crossref
|
|
|
|
|
Brasil, Guia para validação de métodos analíticos. Diretoria colegiada da Agência Nacional de Vigilância Sanitária, Resolução No899, 2003.
View
|
|
|
|
|
Brasil, Lista A de medicamentos de referência, 2015, presentation slides available at
View
|
|
|
|
|
Chik Z, Tucker AT, Shiel JI (2010). Comparative Pharmacokinetic Assessments of Topical Drugs: Evaluation by Dermatopharmacokinetics, Microdialysis, and Systemic Measurement. J. INVEST. DERMATOL. 130(12): 2828-2830.
Crossref
|
|
|
|
|
FDA Guidance for industry on special protocol assessment (2002). Fed. Regist. 67:35122.
|
|
|
|
|
Hathout RM, Mansour S, Geneidi AS, Mortada ND (2011). Visualization, dermatopharmacokinetic analysis and monitoring the conformational effects of a microemulsion formulation in the skin stratum corneum. J. colloid interface sci. 354(1):124-130.
Crossref
|
|
|
|
|
Herkenne C, Naik A, Kalia YN, Hadgraft J, Guy RH (2007). Ibuprofen transport into and through skin from topical formulations: in vitro–in vivo comparison. J. Invest. Dermatol. 127(1):135-142.
Crossref
|
|
|
|
|
Herkenne C, Naik A, Kalia YN, Hadgraft J, Guy RH (2006). Dermatopharmacokinetic prediction of topical drug bioavailability in vivo; Pharm. Res. 8(4): 887-894.
|
|
|
|
|
Lodén M, Åkerström U, Lindahl K, Berne B (2004). Bioequivalence determination of topical ketoprofen using a dermatopharmacokinetic approach and excised skin penetration. Int. J. pharm. 284(1):23-30.
Crossref
|
|
|
|
|
Patel NA, Patel NJ, Patel RP (2009). Drug Discov. Ther. 3(5):234-242.
|
|
|
|
|
N'Dri-Stempfer B, Navidi WC, Guy RH, Bunge AL (2008). Optimizing metrics for the assessment of bioequivalence between topical drug products. Pharm. Res. 25(7):1621-1630.
Crossref
|
|
|
|
|
Nagelreiter C, Mahrhauser D, Wiatschka K, Skipiol S, Valenta C (2015). Int. J. Pharm.491:162-169.
Crossref
|
|
|
|
|
Pershing LK, Bakhtian SC, Poncelet J, Corlett EL, Shah VP (2002). J. Pharm. Sci. 91:5.
Crossref
|
|
|
|
|
Pershing LK, Corlett J, Jorgensen C (1994). In vivo pharmacokinetics and pharmacodynamics of topical ketoconazole and miconazole in human stratum corneum. Antimicrob. agents chemother.38(1):90-95.
Crossref
|
|
|
|
|
Dai W, Wang C, Yu C, Yao J, Sun F, Teng L, Li Y (2015). Preparation of a mixed-matrix hydrogel of vorinostat for topical administration on the rats as experimental model. Eur J. Pharm. Sci. 78:255-263.
Crossref
|
|
|
|
|
Wan T, Xu T, Pan J, Qin M, Pan W, Zhang G, Xu Y (2015). Microemulsion based gel for topical dermal delivery of pseudolaric acid B: In vitro and in vivo evaluation. Int. J. Pharm. 493(1):111-120.
Crossref
|
|
|
|
|
Wiedersberg S, Dermatopharmacokinetics and pharmacodynamics of topical glucocorticoids. Thesis submitted for a degree of Doctor of Philosophy, University of Bath, Department of Pharmacy and Pharmacology.
|
|