ABSTRACT
Thaumastocoris peregrinus is a pest that damages eucalyptus plantations. Recently in Brazil, there are few studies related to its control. With the advancement of molecular biology, several techniques can assist in the discovery of an effective and sustainable control. The different techniques of analysis of gene expression start with the extraction of the total RNA from the material for genomic analysis, hence it is essential that the RNA be intact. Numerous insect RNA extraction protocols are available, but none of them are widely effective, therefore, several factors and intrinsic characteristics of the tissue to be analyzed can affect the quality of the extracted material. Thus, it is necessary to modify the protocols in order to optimize the extraction of RNA. This study aimed to analyze the efficiency of three methods for RNA extraction from T. peregrinus. The results obtained demonstrated that the RNA extracted with only one method was more efficient due to less contamination with DNA and greater integrity of the samples. It should be noted that the determination of an efficient protocol for the extraction of RNA from T. peregrines will assist in future research, which will assist in the discovery of genes present in the extracted RNA sample.
Key words: Ribonucleic acid, bronze bug, Eucalyptus.
Eucalyptus production is becoming increasingly prominent in the Brazilian economy. Countless advances have been achieved in terms of productivity and the total planted area has been growing. However, since their introduction in the country for commercial purposes, eucalyptus species have suffered from the attack of pests and diseases. Among these, the bronze bug, Thaumastocoris peregrinus (T. peregrinus) Carpintero and Dellapé, 2006 (Hemiptera: Thaumastocoridae), was detected in the year. 2008. The bronze bug is an exotic pest and has since attacked eucalyptus plantations, reducing its production. This insect pest has adapted and spread throughout the Brazilian territory, starting to cause direct damage to eucalyptus plants, a fact that caused a considerable increase in production costs and generated serious commercial losses (Wilcken et al., 2010).
As a phytophagous species, when feeding, the tan bug perforates the leaves and fine branches to suck the sap, causing a silvering of the leaves and, later, the leaves look tan and suffer abscission, with consequent reduction of photosynthetic area of the plant. In case of severe attacks, it can lead to death (Jacobs and Neser, 2005).
When it sucks the leaf sap, it causes severe damage due to the possible release of enzymes, which leaves the leaf with a tanned appearance, reducing the photosynthetic area of ​​the plant. The identification and characterization of the molecular mechanisms of the insect responsible for leaf damage are important to support population control strategies for T. peregrinus, mainly through the resistance of plants. Molecular biology techniques favor this approach and in this way, this work aims to identify and characterize the expression of important genes that are related to leaf damage caused by the insect.
However, the use of these molecular techniques depends directly on obtaining good quality nucleic acids including RNA in adequate quantities. Studies in the field of molecular biology have evolved rapidly and new techniques have been shown to be useful in the quantification of expression patterns. However, the numerous techniques of analysis of gene expression currently available directly require obtaining good quality and adequate quantities of the extraction of pure and intact RNA (Aras et al., 2003). These steps are essential for a comparative quantitative analysis.
Extraction procedures are not absolutely reproducible for all species, since they possess different types of tissues and cells (Gouveia and Regitano, 2007). In addition, it is essential to accumulate a significant amount of RNA for some procedures, which can become complicated to small insects, such as the bronze bug (T. peregrinus).
In practice, the procedures are empirical, as it is essential to making adaptations and modifications to the protocols (Chiari et al., 2009); also, it is essential to use particular methodologies in order to optimize the extraction of RNA from a good quality sample (Waldschmidt et al., 1997). Furthermore, the protocol in use must be appropriate to the objective that will be given to the extraction of the nucleic acid, such as quantitative real-time PCR (qRT-PCR), sequencing, cloning, gene expression, etc., to find results conducives and acceptable to the application (Bartlett and Strirling, 2003).
RNA extraction and isolation consists of the initial lysis and cell denaturation phase in which cell membranes are disrupted and total nucleic acids extracted. The predominant difficulty in RNA extraction is its degradation by the present stable and active ribonucleases (RNases) in the tissues (Bitencourt et al., 2011). Therefore, initial extraction buffers must have RNase inhibition to prevent the action of RNases (Romano and Brasileiro, 1999). The second stage consists of the separation of RNA from other unwanted cellular components, such as membrane debris, proteins and DNA and the third and last stage, involves precipitation of RNA, usually with the use of alcohols (ethanol or isopropanol) (Gouveia and Regitano, 2007).
With the premise of ensuring high quality RNA extraction from small amounts of biological material, numerous commercial kits are manufactured, and for the most part, they are in fact efficient; however, it is essential to observe the viability for the species and tissue you want to work on (Bitencourt et al., 2011). As an alternative, different protocols and reagents for the extraction of RNA from arthropods have been described in several studies; however, for the extraction of T. peregrinus specifically, they are not efficient. We were able to extract the RNA, but for use after extraction it is not satisfactory.
Considering that it is not only in places where T. peregrinus has been introduced that there is a need to generate and make available information that helps in the management of this insect pest, in a bid to reduce economic losses and guarantee the productive and environmental sustainability of the stands of Eucalyptus, as well as the the difficulty in sources of resistance for genetic improvement of the eucalyptus for the pest in focus and the limitations in the efficiency and durability of chemical treatments and biological control, a promising alternative method of control such as gene silencing by interfering RNA (RNAi) can be developed. The genomic and transcriptomic sequences of T. peregrinus necessary for the selection of target genes are still unknown. With focus on sequencing the transcriptome of the bronze bug, the present study aimed to establish protocols for obtaining the extraction of total RNA from T. peregrinus samples.
The adult insects of T. peregrinus were collected in the field at the Campus II of the Federal University of the Jequitinhonha and Mucuri Valleys, located in the city of Diamantina-MG. Subsequently, 5 insects were released in 5 Petri dish for each protocol (15 Petri dish) with 5 repetitions for each protocol (Figure 1), where they were fasted for 2 h, then insects from Petri dish were transferred to an Eppendorf tube and immediately frozen in liquid nitrogen, in order to maintain the integrity of the samples, facilitate the extraction processes and keep the metabolic conditions related to the attack process of T. peregrinus stable; thereafter, we proceeded to extract the RNA immediately.
Three protocols used to extract total RNA from insects were used in the experiment and are described below.
All procedures were adopted with the following cleaning / sterilization measures to ensure that the site was free of any contaminants:
1) The solutions used were made with DEPC water (diethylporocarbonate) properly autoclaved;
2) All glassware and other utensils were previously autoclaved at 121°C for a period of 30 min and then dried in an oven;
3) Polypropylene tips and microtubes are new and free of RNAs;
4) The bench used is exclusively for the handling of RNA and was constantly cleaned with SDS 10% and with 70% ethanol, as were all the equipment used in the experiment;
5) In addition, the necessary care was taken to keep the gloves used free of RNAs;
6) The experiments were carried out in a controlled environment at a temperature of 23°C.
Protocol 01: TRI Reagent ® - SIGMA with modifications described
In centrifugation microtubes (Eppendorf) of 1.5 ml, 5 insects were deposited along with 1000 µl of trizol® buffer (Sigma); thereafter 3 microspheres (beads) were added to the tube. With the aid of a mechanical disruptor (Mini-Beadbeater ™), the total RNA was extracted. This done, the samples were incubated for 5 min on ice, then 200 µl of chloroform were added and the microtubes were shaken for 15 s via manual inversion, and promptly incubated for another 15 min on ice. Subsequently, the samples were centrifuged for 15 min at 4°C and 10351 xg. The next stage consisted of washing the material and for this, the supernatant was transferred to a new tube, 500 µl isopropanol was added, the solution was mixed via inversion and incubated for 10 min on ice. Further, the sample was subjected to another centrifugation for another 10 min at 4°C and 10351 xg. In this step, the supernatant was removed so that only the precipitated material (Pellet) remained and immediately 1000 µl of 75% ethanol was added, mixed via inversion for approximately 15 s and again centrifuged, this time for 5 min at 4°C and 8000 RPM.
The supernatant was again discarded and the precipitated material dried for 10 min in a laminar flow chamber. To the dry material, 35 µl of water treated with the diethyl pyrocarbonate reagent (DEPC) was added, as it is a strong inhibitor of RNase activity.
Protocol 02: D-Sorbitol – SIGMA
In centrifugation microtubes (Eppendorf) of 1.5 ml, 3 microspheres (beads) were added together with up to 5 insects and stirred in the Disruptor (Mini-Beadbeater ™). Thereafter, 250 µl of the D-Sorbitol wash buffer and 15 µl of 2-βmercaptaethanol (β-ME) per sample were added and mixed via stirring in the Disruptor (Mini-Beadbeater ™) for 10 s. Subsequently, the tubes were centrifuged for a period of 5 min at 5000 xg.
Further, the supernatant was discarded and the washing process repeated for at least one more time, or until the buffer shows no suspension. In this step, 120 µl of cetyltrimethylammonium bromide (CTAB), preheated to 65°C, together with 7 µl of β-ME per sample were added, the solution was homogenized by stirring with the aid of the Disruptor (Mini-Beadbeater™) for 10 s and then incubated in a water bath at 65°C for 60 min, with homogenization by manual inversion every 10 min.
Subsequently, the material was cooled on ice for 5 min, and after that, 700 µl of chloroformizo: isoamyl alcohol (CIA 24: 1) was added, followed by stirring for 10 s in the Disruptor (Mini-Beadbeater ™) and centrifugation at 5,000 xg for 10 min at 4°C. About 600 µl of the supernatant was promptly transferred to a new microtube, 700 µl of CIA was added, the solution was stirred again for 10 s in the Disruptor (Mini-Beadbeater ™) and subjected to centrifugation for 10 min at 4°C and 13000 xg.
After this step, the supernatant was transferred to another microtube, in addition to 1/10 of the volume of 3 M sodium acetate, pH 5.2 and 1/3 of the volume of cold isopropanol (isopropyl alcohol), the solution was mixed via inversion and the tubes stored at -20°C for a minimum period of 1 h.
Centrifugation was carried out at 13,000 xg for 10 min at 4°C. The supernatant was removed so that only the precipitated material was left (Pellet) and immediately 1000 µl of 70% ethanol was added. Again, centrifugation was carried out at 13,000 xg for 10 min at 4°C. Supernatant was again discarded and the precipitated material dried for 10 minutes in a laminar flow chamber.
Protocol 03: RNeasy Mini-kit – Qiagen
Before starting the extraction itself, the following was performed:
1. Addition of 10 µl of β-mercaptoethanol (β-ME) or 20 µl of 2 M dithiothreitol (DTT) to 1 ml of RLT buffer.
2. Addition of 4 volumes of ethanol (96–100%) to Buffer RPE for a working solution.
A maximum of 30 mg of tissue (indicated by the manufacturer) was macerated with BUFFER RLT and then the appropriate volume of Buffer RLT was added, as described in Table 1. The tubes were centrifuged for a period of 3 min at a speed of 13,000 xg.
Furthermore, the supernatant was carefully removed with the aid of a pipette, 1 volume of 70% ethanol was added to the lysate and again the solution was homogenized with the aid of the pipette; also, the manufacturer did not recommend centrifugation. For this step, it was necessary to transfer 700 µl of the sample, including any precipitate, to an RNeasy Mini column spin previously placed in a 2-ml collection tube (provided in the kit) and the material was centrifuged for 15 s at ≥ 8000 xg. At the end, the tube was discarded, 350 µl of RW1 buffer was added to the RNeasy column and the samples were centrifuged for 15 s at ≥ 8000 xg. The residual solution was discarded and the column returned to the tube.
Concomitantly, 10 µl of DNase I stock solution (obtained by adding 550 µl of RNase-free water and homogenizing by manual inversion) was added to 70 µl of RDD buffer. The solution was gently homogenized by inversion and spun briefly. That done, 80 µl of the DNase incubation mixture was added directly to the RNeasy column membrane and the samples were incubated at room temperature for 15 min.
Subsequently, 350 µl of RW1 buffer was added to the RNeasy column, the solution was centrifuged for 15 s at ≥ 8000 xg and the flow was discarded. The RNeasy column was returned to the tube and 700 µl of RW1 buffer was added, with subsequent centrifugation for 15 s at ≥ 8000 xg and the flow discarded.
At the end of this period, 500 µl of RPE buffer was added to the RNeasy rotation column, the lid was closed and the solution was centrifuged for 15 s at ≥ 8000 xg; thereafter the flow was discarded. Further, 500 µl of Buffer RPE was added to the RNeasy rotation column, followed by decentrifugation for 2 min at ≥ 8000 xg.
In order to dry the column, centrifugation was performed at 13,000 xg for 1 min. Then, the RNeasy centrifuge column was transferred to a new 1.5 ml collection tube and 50 µl of RNAse-free water was added directly to the column membrane and centrifuged for 1 min at ≥ 8000 xg, to dilute the precipitated RNA.
At the end of each extraction process, the total RNA samples obtained were subjected to integrity analysis using the analytical method of electrophoretic visualization in 1.5% agarose gel, stained with ethidium bromide and visualized under ultraviolet light, in transilluminator, and photo-documented in a photo-documenter (LoccusBiotecnologiaTransluminator L. Pix) coupled to a microcomputer.
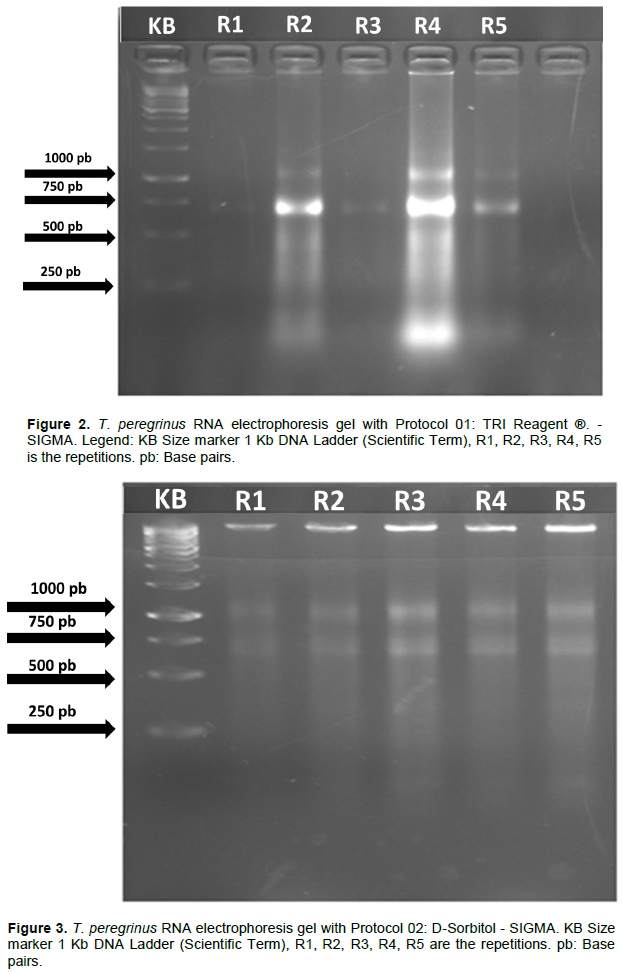
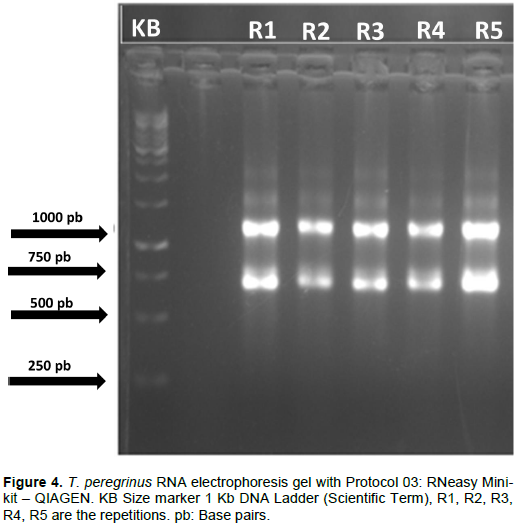
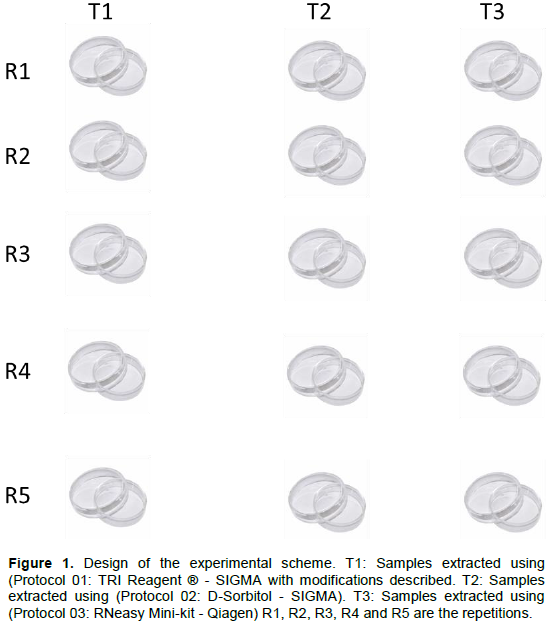
A difference was observed in the efficiency of the three protocols tested regarding the quality and quantity of the extracted RNA. The analysis by electrophoresis allowed verification such that the separation of the bands of the different types of RNAs was easily observed for the three protocols tested in this study (Figures 2 to 4).
However, the extraction performed with the commercial kit “RNeasy Mini-kit” (Qiagen) showed clearer and more intense bands, for all samples analyzed (Figure 4). Although with lower band intensity, extraction with D-Sorbitol (SIGMA) also allowed an invariable standard of total RNA quality for all samples (Figure 3).
Among the tested protocols, only Protocol 01, using the reagent TRIS (SIGMA), showed band intensity variability as a function of the sample used for extraction. No clear bands were observed for samples 1 and 6 (Figure 2). It is also noteworthy that, apart from the gel referring to Protocol 03 (Figure 4), it was not possible to verify the presence of DNA bands being the most suitable for possible future reactions. The results of the electrophoresis reveal that the best protocols for the extraction of total RNA, based on the estimated amount of RNA and the success of the extraction from the different materials, were Protocols 02 and 03 in which all presented RNA with pattern of desired band in the Protocol 03 agarose gel showed visible band and without drag indicating greater integrity of the obtained RNA. A difference was observed in the efficiency of the three protocols regarding the quantity and quality of the RNA extracted for T. peregrinus.
To carry out studies of gene expression and characterization of transcripts, it is necessary that RNA be extracted and purified efficiently, in such a way that its integrity and quality are maintained. Also, the extraction process needs to be safe, easy to perform, low cost, and allows its repeatability (Deng et al., 2005; Ibelli et al., 2007).
The techniques available for RNA extraction are numerous and despite their variations, the basic principle is the lysis of lipid membranes by a detergent solution, followed by purification, precipitation and RNA elution. The exposure of the genetic material to extraction buffers, as well as the initial stage of the process, helps in the precipitation of RNA and in the maintenance of its integrity in the subsequent stages of extraction, through the degradation of endogenous RNases (activities that are possible due to the presence of compounds such as lithium chloride and guanidine thiocyanate) (Sambrook and Russel, 2001).
The various commercially available reagents have combined substances in their compositions, such as guanidine isothiocyanate and phenol, which enable RNA extraction faster than that of conventional protocols and guarantee the integrity of the material. However, it is necessary to use other reagents in the subsequent steps to assist in the sample purification process, such as the chloroform that solubilizes the lipids and allows their removal from the RNA molecules.
An ideal extraction technique should allow obtaining a large amount of pure and intact RNA, be easy and quick to perform, and also reproducible; in addition to being low cost and capable of extracting a large number of samples simultaneously. However, not all available techniques combine all of these characteristics together. Each technique has advantages and disadvantages that must be reviewed before a laboratory decides which one is most suitable for it, which makes works that perform protocol tests according to the tissue and/or species studied, of fundamental importance.
In the present study, the three protocols tested have different extraction buffers, with peculiar characteristics that seek to maintain the integrity of RNA throughout the process of extracting the molecule. The advantage of these reagents is that they are marketed ready for use, the procedure is quick and direct, and they promote rapid denaturation of nucleases and stabilization of RNA.
Among the tested processes, the extraction of RNA with TRIS (SIGMA) is considered one of the simplest and most used ways to extract RNA from cells or tissues. Based on the technique of Chomczynski and Sacchi (1987) and commercial availability, TRI Reagente® offers the possibility of simultaneous extraction of DNA and proteins present in the sample. It is a monophasic solution of phenol and guanidine isothiocyanide that disrupts cells and dissolves cellular components.
Despite presenting the advantages of being a ready-to-use reagent and a fast and direct procedure, capable of promoting rapid RNase denaturation and RNA stabilization, this protocol (Protocol 01) did not present satisfactory results for the extraction of T. peregerinus RNA. The inefficiency in extracting the RNA from the different samples of the insect used in the process was evidenced by the absence of ribosomal RNA bands observed in the image of the agarose gel, for samples 1 and 6, as well as, by the low intensity of the bands for samples 3 and 5 (Figure 2). The presence of sharp and shiny ribosomal RNA bands (28 S and 18 S) in the agarose gel is an indication of the good quality and integrity of the extracted RNA.
The analysis by agarose gel electrophoresis helped to verify also, that the only two samples that presented intense bands (samples 2 and 6), also, presented intense “traces”, that enabled us infer the existence of contamination and possible degradation of the samples. In addition, the presence of the DNA band was found, although not very evident, a fact that is directly linked to Trizol's inability to remove DNA from plasmids and DNA fragments, being able to efficiently remove only the large molecules of this acid nucleic.
In short, despite the homogenization of T. peregrinus samples obtained using the Mechanical Disruptor (Mini-Beadbeater™), equipment that causes cellular disturbances through constant and high-speed impacts and makes the process of homogenizing and extracting RNA extremely efficient in ensuring greater contact with the Trizol reagent, and which, in theory, should guarantee better denaturation of tissue proteins should be used. Also, despite the use of low temperatures in order to interrupt the enzymatic activity of RNases, it was not possible to obtain high-quality RNA samples with the use of the reagent TRI, which proved to be a poorly reproducible technique by allowing the extraction of RNA from only two samples, among the six analyzed.
Extractions performed with reagent IRT, following the manufacturer's recommendations, depending on the tissue and/or species, may have low yield and, consequently, high contamination due to the presence of proteins and phenolic compounds (Jaakola et al., 2001; Martins et al., 2018). As it is a reagent composed of phenol, when used in inadequate reagent/tissue proportions, it can also cause damage to the poly-A tail of the extracted mRNA (Azevedo et al., 2003), which can hinder further analysis. In addition, DNA fragments that are not removed in the extraction process with the reagent TRI may compromise procedures that depend on a high RNA yield such as the polymerase chain reaction via reverse transcriptase (RT-PCR).
In contrast, as with the application of Protocol 02 (D-Sorbitol - SIGMA) in this study, many protocols have been based on modifications of the classic extraction protocol with cetyltrimethylammonium bromide (CTAB) (Doyle and Doyle, 1987) to improve the purity and yield of DNA and RNA extraction. Among these modifications, the use of a sorbitol-based solution to pre-wash the tissue to be analyzed has allowed a reduction in the amount of extracellular contaminants and the obtaining of high quality and integrity RNA samples (Chang et al., 1993) with samples of different tissues and species.
Although, the use of D-Sorbitol in the present study promoted obtaining integral RNA samples and, apparently, free of polyphenols and polysaccharides, it can be inferred based on the absence of “traces” and the sharpness of the bands in the image of the agarose gel (Figure 3). The low intensity of the observed bands also shows a low amount of extracted RNA, which may be undesirable depending on the technique of analysis of gene expression subsequently employed. Another point to be highlighted refers to the presence of intense DNA bands observed in all samples processed with Protocol 02.
Thus, the electrophoresis analysis of the agarose gel showed that the use of D-Sorbitol together with 2-ßmercaptoethanol, which helps to reduce the oxidation of RNA pellets, constitutes a reproducible technique and provides RNA samples with high integrity. However, adjustments are necessary to obtain a higher concentration of RNA at the end of the process, alongside a subsequent treatment with the enzyme DNase I in order to remove fragments of genomic DNA. In comparison, Protocol 03 (RNeasy Mini-kit - Qiagen) was the only procedure that managed to combine the integrity, quantity and quality of the extracted RNA with the repeatability of the extraction technique used. This allows the use of these samples in subsequent methods of analysis that involve, for example, enzymatic digestion, amplification and sequencing.
The use of extraction kits is based on RNA adsorption. They use the property of this nucleic acid to bind on surfaces such as magnetic spheres, silica, polystyrene latex materials, cellulose matrix or glass fibers, in the presence of certain salts or chaotropic agents that have the property of disrupting the three-dimensional structure of nucleic acids and proteins, by denaturing these macromolecules which allows high yield from small amounts of biological tissue.
RNA samples with these qualities, so desirable, are possible due to the use of the column that accelerates, facilitates, guarantees the extraction profit and reduces the risk of contamination during the process (Fanson et al., 2000; Siddappa et al., 2007). This works together with the use of the stabilizing solution RNAlater® that quickly permeates most tissues to stabilize and protect the RNA and slow down its degradation process, which is essential in the extraction process, as well as the addition of the enzymatic treatment with DNase I, which allows, as seen in the image of the agarose gel (Figure 4), the complete removal of the DNA fragments.
In contrast, the use of column kits for RNA extraction has the main disadvantage of the high price. In addition to not being a suitable technique for the extraction of small RNAs, some researchers also describe that it can result in the extraction of less pure RNA compared to organic extraction (Fanson et al., 2000; Siddappa et al., 2007).
Accordingly, it is understood that there is no nucleic acid extraction protocol (DNA or RNA) that can be considered standard. The presence of polysaccharides, polyphenols and a wide range of secondary metabolites makes it difficult to standardize between different tissues and obtain high-quality RNA (Campos et al., 2010; Cardillo et al., 2006) having all protocol advantages and disadvantages that must be carefully analyzed after the use of tests directed to the material under study. Each biological tissue or species of organism has characteristics intrinsic to its chemical and structural composition that can interfere with the success of the different stages of the extraction process. At the same time, each reagent used has its specificity and importance in this process, aimed at contributing to the isolation, purification and/or integrity of the RNA molecule. This makes it possible and important to test and manipulate the different protocols available for extraction, when starting a molecular study with a species such as T. peregrinus.
The commercial kit “RNeasy Mini-kit” (Qiagen) was the most efficient method for extracting total RNA from T. peregrinus. However, the extraction protocol with the regent D-Sorbitol (Sigma) has also shown satisfactory results and can actually replace the Kit, provided new tests are carried out that allow a greater yield in the extraction process with this reagent. On the other hand, extraction with the use of reagent Tris is not recommended due to its low effectiveness.
The authors have not declared any conflict of interests.
The authors are grateful to Aperam Bioenergy LTDA, FAPEMIG, CNPq, CAPES and UFVJM for logistical and financial support to carry out this work.
REFERENCES
|
Aras S, Duran A, Yenilmez G (2003). Isolation of DNA for RAPD analysis from dry leaf material of someHesperis L. specimens. Plant Molecular Biology Reporter 21(4):461-462
Crossref
|
|
|
|
Azevedo H, Lino-Neto T, Tavares RM (2003). An improved method for high quality RNA isolation from needles of adult maritime pine trees. Plant Molecular Biology Reporter, Athens 21(4):333-338.
Crossref
|
|
|
|
|
Bartlett JMS, Strirling D (2003). PCR protocols. 2. ed. New Jersey: Humana Press. 226.
Crossref
|
|
|
|
|
Bitencourt GDA, Chiari L, do Valle CB., Laura VA, Moro JR (2011). Avaliação de diferentes métodos para extração de RNA total de folhas e raízes de braquiária. Campo Grande, MS: Embrapa Gado de Corte Boletim de Pesquisa e Desenvolvimento (INFOTECA-E) 29(1):1-26.
|
|
|
|
|
Campos NA, Alves JD, Porto BN, Souza KRD, Santos MO, Silveira HRO, Magalhaes MM (2010). Otimização de protocolos de extração de RNA de raiz de milho (Zea mays) visando estudos moleculares de alta sensibilidade. In: CONGRESSO NACIONAL DE MILHO E SORGO, Goiânia. Anais Goiânia: Associação Brasileira de Milho e Sorgo, 2010. 28:97-104.
|
|
|
|
|
Cardillo AB, Giulietti AM, Marconi PL (2006). Analysis and sequencing of h6hmRNA, last enzyme in the tropane alkaloids pathway from anthers and hairy root cultures of Brugmansia candida (Solanaceae). Electronic Journal of Biotechnology, Cambridge 3:196-198.
Crossref
|
|
|
|
|
Carpintero DL, Dellapé PM (2006). A new species of Thaumastocoris Kirkaldy from Argentina (Heteroptera: Thaumastocoridae: Thaumastocorinae). Zootaxa 1228:61-68.
Crossref
|
|
|
|
|
Chang S, Puryear J, Cairney JA (1993). Simple and Efficient Method for Isolating RNA from Pine Trees. Plant Molecular Biology Reporter 11(2):113-116.
Crossref
|
|
|
|
|
Chiari L, Valle JVR, Resende RMS (2009). Comparação de três métodos de extração de DNA genômico para análises moleculares em Stylosanthes guianensis. Embrapa Gado de Corte, Circular Técnica 36, 1a edição.
|
|
|
|
|
Chomczynski P, Sacchi N (1987). Single-step method of RNA isolation by acid guanidiniumthiocyanate-phenol-chloroform extraction. Analytical Biochemistry 162(1):156-159.
Crossref
|
|
|
|
|
Deng MY, Wang H, Ward GB, Beckham TR, Mckenna TS (2005). Comparison of six RNA extraction methods for the detection of classical swine fever virus by real-time and conventional reverse transcription-PCR. Journal of veterinary diagnostic investigation: Official Publication of the American Association of Veterinary Laboratory Diagnosticians, Inc.
Crossref
|
|
|
|
|
Doyle JJ, Doyle JL (1987). Arapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19:11-15.
|
|
|
|
|
Fanson BG, Osmack P, DI Bisceglie AM (2000). A comparison between the phenolchloroform method of RNA extraction and the QIAamp viral RNA kit inthe extraction of hepatitis C and GB virus-C/hepatitis G viral RNA from serum. Journal of Virological Methods 89(1-2):23-27.
Crossref
|
|
|
|
|
Gouveia JJS, Regitano LCA (2007). Protocolos de Biologia Molecular aplicadas à Produção Animal. São Carlos: Embrapa 72.
|
|
|
|
|
Ibelli AMG, Regitano LC, de A, Nicimura SCM (2007). Extração de RNA. In: Regitano, L. C. De A.; Niciura, S. C. M.; Ibelli, A. M. G.; Gouveia, J. J. De S. Protocolos em biologia molecular aplicada à produção animal. São Carlos: Embrapa Pecuária Sudeste 1:9-13.
|
|
|
|
|
Jaakola L, Pirttila AM, Halonen M, Hohtola A (2001). Isolation of high quality RNA from bilberry (Vacciniummyrtillus L.) fruit. Molecular Biotechnology, Washington 19 (2):201-203.
Crossref
|
|
|
|
|
Jacobs DH, Neser S (2005). Thaumastocoris australicus Kirkaldy (Heteroptera: Thaumastocoridae): a new insect arrival in South Africa, damaging to Eucalyptus trees: research in action. South African Journal of Science Pretoria 101(5):233-236.
|
|
|
|
|
Martins JC, Silva JRNJ, Almeida PCR, Vilanova-Costa CAST, Saddi VA (2018). Padronização e Comparação de Métodos Aplicados à Extração de RNA Total em Serpentes do Gênero Micrurus. Revista Estudos- Zootecnia.
View
|
|
|
|
|
Romano E, Brasileiro ACM (1999). Extração de DNA de plantas. Biotecnologia 2(9):40-43.
|
|
|
|
|
Sambrook J, Russel DW (2001). Molecular cloning - a laboratory manual. 3. ed. Cold Spring Harbor Laboratory Press: New York.
|
|
|
|
|
Siddappa NB, Avinash A, Venkatramanan M, Ranga U (2007). Regeneration of comercial nucleic acid extraction columns without the risk of carryover contamination. BioTechniques 42(2):186-192.
Crossref
|
|
|
|
|
Waldschmidt AM, Salomão TMF, Barros EG e Campos LAO (1997). Extração de DNA genômico de Melipona quadrifasciata (Hymenoptera, Apidae, Meliponinae). Brazilian Journal of Genetics 20:421-423.
Crossref
|
|
|
|
|
Wilcken CF, Soliman EP, Nogueira de Sá LA, Barbosa LR, Dias TKR, Ferrera-Filho PJ, Oliveira RJR (2010). Bronze bug Thaumastocoris peregrinus Carpintero and Dellapé (Hemiptera: Thaumastocoridae) on Eucalyptus in Brazil and its distribution. Journal of Plant Protection Research 50(2):201-205.
Crossref
|
|