A new method of screening type IIG restriction modification (RM) enzyme has been developed using REBASE, a database of all known and putative restriction enzymes and methyltransferases found throughout the bacterial genome sequences available in GENBANK. The in silico analysis of a group of putative type IIG RM enzymes in Microcystis aeruginosa showed a high sequence homology at both ends. This peculiarity allows for primers designing that can be used in polymerase chain reaction (PCR) to amplify the corresponding genes out of one environmental DNA extracted from a cyanobacteria-rich sample. PCR products were cloned into the pSAPV6 vector. Among eight recombinant DNA sequenced, five showed different sequences in the protein regions that interact specifically with DNA.". These five recombinant proteins expressed type IIG RM enzyme activity. Their specificities were determined, and all correspond to new DNA recognition sites.
The type II restriction enzymes discovered in 1968 (Smith and Welcox, 1970) are endonucleases that cut DNA at specific 4-8 nucleotide sequences which mainly exist in prokaryotes. Each restriction enzyme always pairs with a methyltransferase which modifies host DNA at the same site. The two enzymes form a restriction modification (RM) system that probably contributes to protecting bacteria against foreign DNA (Raleigh and Brooks, 1998). Restriction enzymes are widely used as tools (Roberts, 2005) in molecular biology procedures.
Approximately 4000 restriction enzymes have been characterized, which recognize 365 different sites (Pingoud et al., 2014), thus representing a statistically minor fraction of all the possible DNA sequences. Such a small diversity of specificities may be explained by the first screening procedures that allow analysis of only a small percentage of bacteria able to grow on common rich media. Though sharing the same function of cutting DNA after binding to specific recognition sites, these classical restriction enzymes of different specificities, has no sequence homology that could be used to identify others in bacterial genomes (Wilson, 1991). Their possible location in a genome could only be suggested by the presence of methyltransferase genes (Pósfai et al., 1989) always located close to the related restriction enzyme genes. The lack of sequence homology also makes the protein-DNA interactions impossible to study. The further discovery of restriction enzyme families with high homology sequences, though able to recognize different recognition sites, has facilitated the understanding of protein-DNA interactions in this class of enzymes.
So far, the MmeI (Morgan et al., 2009) and the Thermus families (Skowron et al., 2003, 2017; Zylicz-Stachula et al., 2012) have been discovered. These enzyme families are referred to as Type II C/G enzymes, meaning that restriction and methyltransferase activities are on the same protein, thus a complete RM system in itself, instead of being a heterodimer as for classical RM enzymes. Due to this peculiarity, whenever a mutation occurs altering the nucleotide sequence of the recognition site, the two functions can still operate in a concerted manner by cutting the new nucleotide sequence of foreign DNA and modifying the same new sequence of the host genome. The MmeI family, first discovered, was found through in silico investigations in REBASE (Roberts et al., 2015), a database of real or putative restriction enzymes and methyltransferases screened in all the bacterial genomes available in GenBank. Several putative MmeI-like proteins which have extensive sequence homology were amplified from the original sequenced bacterial DNA and cloned. Active recombinant proteins have been characterized for their recognition specificities. In the two families so far described, the single-chain proteins are similarly structured with the COOH-restriction enzyme domain linked to the methyl transferase domain by a helical domain followed by the target recognition domain (TRD) - NH2. TRD is a variable region which interacts with distinctive DNA sequences (Klimasauskas et al., 1991). Thus, the multi-specific MmeI family has allowed understanding the interaction rules between the amino acids (AA) in the TRD regions and the nucleotides of the recognition sequence. Based on this, the authors were able to modify the enzyme specificity as wished (Morgan and Luyten, 2009). In the Thermus aquaticus family, TaqII and TaqIII have highly similar protein sequences although they have different specificities. These enzymes are clear examples that specificity evolution occurs naturally (Furuta et al., 2010; Furuta and Kobayashi, 2012).
The type IIG RM enzymes can frequently be found in the sequence of bacterial genomes, a finding suggesting they could be an efficient strategy for prokaryotes to diversifying their defensive systems (Blow et al., 2016). In REBASE, Microcystis aeruginosa NIES-843 strain (Kaneko et al., 2007) was found to be particularly rich in such enzymes and especially possessed the Mae843ORF8180 coding for a putative IIG RM enzyme. A BLAST search using this enzyme as query, identified several putative genes in many different strains of M. aeruginosa. Strikingly, the translated proteins possessed highly conserved sequences at both -COOH and -NH2 ends. In contrast, the TRD region about 1200 nucleotides long is variable. This observation led us to screen for the presence of similar enzymes in environmental DNA, using both ends as primers for polymerase chain reaction (PCR) amplification of the corresponding genes. M. aeruginosa is blue-green algae widespread in countries such as Vietnam (Duong et al., 2013, 2014), present in blue-green waters where they can overgrow and form a green film at the surface of ponds. This article describes the characterization of type IIG RM genes in environmental DNA extracted from a pond at Cau Dien, Hanoi, encoding proteins bearing novel recognition sites.
Natural starting material
A pond located at Cau Dien, Nam Tu Liem, Hanoi was selected for its green color water. A water sample of around 200-300 ml was taken out of it. After 1 g sedimentation of large debris in the sample bottle, 200 ml of the supernatant were centrifuged for 8 min, at 3100
g in 50 ml
Falcon tubes to pellet down living cells. The pellet was transferred into a 1.5 ml Eppendorf tube and washed 3 times in TE (10 mM Tris, 1 mM EDTA). Aliquots of 50 µl pellets were stored at -30° C. Upon microscopic control, bacterial mass cells characteristic of
M. aeruginosa were observed.
Environmental DNA extraction
The environmental DNA is extracted from thawed 50 µl pellets using DNeasy Plant mini kit (
Qiagen) (Schober and Kurmayer, 2006). DNA is eluted in 100 µl TE. DNA concentration as estimated on agarose gel with a standard DNA marker, is about 20 ng/µl.
Detection of Mae843ORF8180 - like genes in environmental DNA by PCR
Primers were produced by Integrated DNA Technologies,
Inc. The forward primer has been designed so as to anneal at the 5’ region of the
Mae843ORF8180 gene with an extension of the PstI and NdeI restriction recognition sites to facilitate further cloning of PCR products in appropriate plasmids: 5'GTTCTGCAGTTAAGGTTTAACATATGTCTAGATTATTAATCAGCCAGTATCAG3'. The reverse primer anneals to the 3’end of the gene with the BglII recognition site: 5'GTTGTTAGATCTTTAATGTCTCATCGCTTCTATTATTTTCAT3'. PCR was
performed using 1 µl of environmental DNA at 4 different MgCl
2 concentrations (1.5; 2.5; 3.5 and 4.5 mM). Reaction volume is 60 µl composed of 0.02 U/µl Q5 Hot Start polymerase (New England
Biolabs), Buffer Q5 polymerase 1X, 200 µM dNTP; 0.2 µM forward primer and reverse primer. The PCR conditions are: one initial cycle at 98°C, 30 s, followed by 30 cycles of 98°C for 10 s, 63°C for 20 s, 72°C 1 min 30 s and final elongation at 72°C for 2 min. PCR products obtained at all 4 different MgCl
2 concentrations, are mixed and further concentrated and purified on Zymo 25 column (Zymo Research, USA).
Gene cloning
Purified PCR products (200 µl) were cut with 2 µl NdeI and 3 µl BglII (NEB and then purified on Zymo 5 column (Zymo research, USA). The restricted fragments were ligated into the pSAPv6 T7 expression vector (Samuelson et al., 2004) (provided by New England
Biolabs). The recombinant plasmids were used to transform
Escherichia coli ER3081 (F
-l
- fhuA2 lacZ::T7 gene1 [lon] ompT gal attB::(pCD13-lysY, lacI q)
sulA11 R(mcr-73::miniTn10–TetS)2 [dcm] R(zgb-210::Tn10 –TetS) endA1D(mcrC-mrr)114::IS10) provided by New England
Biolabs. Colonies were tested for the presence of the gene by PCR as follows. Cells of individual bacterial colonies were put into 100 µl distilled water and heat broken at 100°C, 5 min. One µl of the resulting solution was assayed with
Quick-Load Taq 2X Master Mix in 30 µl reaction volume. PCR conditions were: one initial cycle at 95°C for 30 s, followed by 30 cycles of 95°C for 15 s, 53°C for 30 s, 68°C for 3 min and a final elongation at 68°C for 5 min. Positive cells were grown overnight and recombinant plasmids were extracted from 3 ml cell cultures using Qiagen Miniprep Kit.
Nested PCR to amplify TRD regions
A nested PCR is carried out to amplify the 1190 bp long TRD variable regions lying between the nucleotide 1326 and 2507 of the
Mae843ORF8180 gene. The forward primer 5’- ATTGGGAATCCTCCTTATAATGCT-3’ and the reverse primer 5’-GTAGTGGAAGATGTCGAGTTTGGT-3’, were used to amplify 40 ng recombinant plasmid with Taq polymerase under the following conditions: one initial cycle at 95°C for 30 s, followed by 25 cycles of 95°C for 15 s, 48°C for 30 s, 68°C for 1 min 15 s and a final elongation at 68°C for 5 min. The amplified TRD regions were then sent to
VNDAT Co. Ltd. for sequencing.
Recombinant protein expression
Recombinant cells were picked up and analyzed as follows for the presence of a specific endonuclease activity. Each single colony was grown in 30 ml LB + chloramphenicol medium at 37°C for about 3 h on a high speed rotation shaker, till reaching exponential growth. Gene expression was induced by adding 0.3 mM Isopropyl-β-D-1-Thio galactopyranoside (IPTG,
Sigma) and the culture was prolonged for 2 more hours. Cells then after were centrifuged at 4°C, at 3100
g and the pellet resuspended in 1.5 ml sonication buffer (20 mM Tris, 1 mM DTT, 0.1 mM EDTA). The pellets were frozen at -30°C and thawed before being lyzed with 20 µl lysozyme 10 mg/ml, 1 h at 4°C. lyzed cells were centrifuged at 12000
g (4°C) and the supernatant was assayed for restriction activity in a 25 µl reaction volume containing Cutsmart buffer, S-Adenosylmethionine (New England
Biolabs) and 0.3 µg pAde2-BsaBI standard DNA [Adenovirus-2 (GenBank Accession #: NC_001405), cut with the restriction enzyme BsaBI (position 4051 and 23479) and ligated into pUC19. In some cases, restriction activity was stronger after a fractionation step on a 1 ml Heparin Sepharose column washed in sonication buffer and a 50 mM-0.9 M NaCl gradient elution. The restriction patterns were analyzed on a 1% agarose gel. Specific methyltransferase activity was detected by the
SMRT sequencing of the recombinant
Escherichia coli genome, performed by New England Biolabs.
Bioinformatics
REBASE was used for analyzing type IIG RM enzymes in Microcystis sp. Similarity searches were performed using BLAST programs (NCBI Resource coordinators, 2016). Sequence alignments were performed using PROMALS3D (Pei and Grishin, 2007).
Screening in REBASE genes coding for type IIG RM putative enzymes from M. aeruginosa
Three putative genes coding for type IIG RM putative enzymes were found in
M. aeruginosa: Mae2549ORF1146,
Mae843ORF8180 and
Mae2481ORF1162. The protein sequences were compared using PROMALS (Figure 1). They had almost the same length, being 997, 998 and 1003 amino acids long, respectively. The sequences at the NH
2- end to AA 412 and at the –COOH end from AA 810 to the end are 100% homologous. These putative RM type IIG enzymes displayed, as in MmeI and Thermus families, three functional domains: i) the Rease catalytic domain extending from AA 1 to AA 117 overpassing the PD-EXK cleavage catalytic motif; ii) the Mtase catalytic domain from AA 303 to AA 451 recognizable by the motif X, GIVYT, the S-Adenosylmethionine binding motif I, LDPTGTGTF, the methylation catalytic motif IV,GNPPY-; and iii) the terminal portion extending from AA 452 includes a variable sequence interacting with DNA target. From AA 118 to AA 302, stands the helical domain which links the REase catalytic domain to the Mtase catalytic domain. A BLASTP search using
one of these protein sequences as a query against the non-redundant Genbank database yielded many results at highly significant expectation values (E equal to 0.0 and identities value > 78% ) in many other proteins of
M. aeruginosa. Analyzing 16 of these proteins, the lengths showed to vary only from 996 to 1011 amino acids and the same conserved sequences were found at both NH
2- and –COOH ends. The
Mae843ORF818 gene was chosen to design primers from 5’ends to be used in PCR experiments on environmental DNA extracted from a natural water sample rich in
M. aeruginosa.

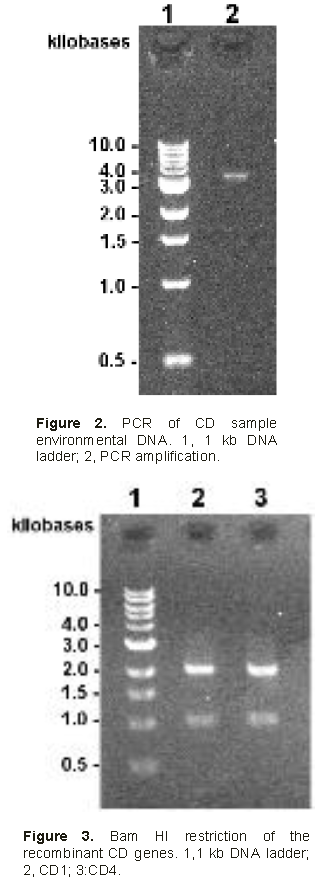
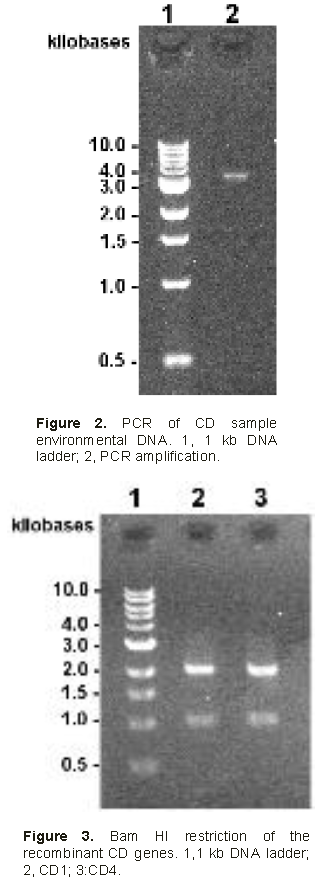
PCR of environmental Cau Dien DNA sample.
The first DNA amplification yielded 3000 bp long products along with much smaller (below 500 bp) non-specific amplification products. The 3000 bp PCR products were purified on agarose gel, cleaned on
Zymo column before being amplified by additional 12 PCR cycles (Figure 2). Overall amplified DNA was estimated to be 120 ng.
Cloning PCR products and identifying recombinant gene.
The cleaned PCR products were restricted with BglII and NdeI and ligated to pSAPV6. The recombinant plasmids were used to transform E. coli. Among 32 transformed clones tested, 8 harbored the expected 3000 bp long insert fragments and were named: CD1, CD4, CD5, CD7, CD10, CD16, CD18 and CD20 respectively
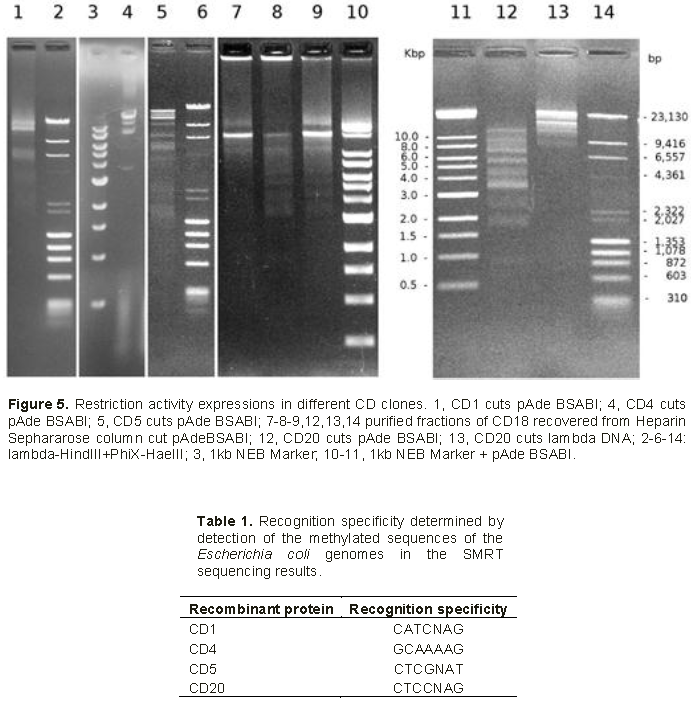
Restriction analysis with BamHI was performed knowing that members of the family of genes under study all have a BamHI site at the nucleotide 996, located in the first conservative region of the coded protein. Figure 3 illustrates the presence of 2 bands as expected at 1000 and 2000 bp. Furthermore, nested PCR has been done to amplify the variable part of the gene coding for the TRD region of the type IIG RM recombinant proteins. The primers correspond to the conserved parts located in the vicinity of the variable part. PCR should yield 1190 bp fragments. The results (not shown here) give bands of the expected size for all CD recombinant strains analyzed. Sequences of these PCR fragments show some heterogeneity among CDs, where CD1, CD4, CD5, CD18 and CD20 were different while CD7 and CD16 were 100% similar to CD1, CD10 being 100% similar to CD4. Thus, 5 distinct genes differing in the sequence of the TRD region, have been identified. Sequences were aligned for comparison using PROMALS3D (Figure 4).

Expression of the recombinant proteins
All supernatants of lyzed recombinant cells displayed restriction activity in assays on standard DNA (Figure 5). CD4 has the highest restriction activity that begins to decrease after the supernatant is 27-fold diluted. The sequence recognition site had already been defined as 5’-GCAAAAG-3’/5’-CTTTTGC-3’ (Le and Nguyen, 2017). Results based on specificity of restriction were confirmed by SMRT sequencing of the CD4 recombinant E. coli genome which showed methyltransferase modifications. Restriction activities of CD1, CD5, CD18 and CD20 were weaker thus the specificities were more difficult to define. Nevertheless, SMRT sequencing of CD1, CD5 and CD20 E. coli recombinants showed the effects of methyltransferase specificities (Table 1) that should also correspond to the restriction activities respectively.
Our results concern the screening in one single natural sample of type IIG RM enzymes alike putative ones found in M. aeruginosa through REBASE. A BLAST search has given other very similar proteins from this genus. All these putative proteins have strictly the same sequences at the beginning and the end of the protein that makes possible to design primers for PCR amplification of the genes present in the environmental DNA extracted from the sample water rich in blue green cyanobacteria. The PCR results give DNA amplification of 3000 bp products that correspond to the chosen in silico gene, Mae843ORF8180. After cloning of individual PCR products, two other experiments confirm this assessment: the BamHI restriction patterns fit the presence of the restricted site in the conservative part of the gene; the nested PCR of the variable region coding for the TRD zone of the protein amplify the right length fragments.
The sequences of 8 recombinant E. coli clones show 5 different DNA sequences coding for the TRD regions (CD1, CD4, CD5, CD18 and CD20). All these recombinant clones have shown restriction activities and the enzyme specificities could have been determined through restriction analysis with CD4 clone which have a strong restriction activity (Le and Nguyen, 2017). Otherwise, the enzyme specificities of CD1, CD5, and CD20 have been determined through the methyltransferase activity on the basis of SMRT sequencing of the respective recombinant E. coli clones.
Thus, from in silico putative genes, we get in one natural sample several genes coding for different active proteins. In this case, we are in presence of the same genes showing allelic diversity in the TRD region (Pingoud et al., 2014). All these enzyme specificities are new. As well as the MmeI-like enzymes, found in silico, have all new specificity recognition (Morgan et al., 2009; Le et al., 2015). In their study, the type IIG RM enzymes were analysed from all known bacterial strains. In this study, the type IIG RM enzymes found in silico in Microcystis aeruginosa strains shared highly homology sequences that allowed us to pick up proteins of different specificities in one natural sample. Thus, the RM enzyme families are naturally adapted to change easily their recognition site specificities. Indeed, this is an adequate way for the host bacteria to adjust rapidly against the phages that could have escaped restriction at the current recognition site.
The next step should be the characterization of more Mae843ORF8180- like genes in other local cyanobacteria rich samples. On this basis, predictions of the interactions between the amino-acid and recognized DNA bases could be made in order to be able to engineer these enzymes and generate the desired recognition sequences (Morgan and Luyten, 2009; Callahan et al, 2016). Analysing genes from bulk natural DNA could be more efficient than from in vitro grown cells. This could be useful to find genes in bacteria that live in special environmental conditions that are difficult to reproduce under laboratory conditions, or for bacteria which grow slowly, such as M. aeruginosa cells which requires one week dividing. Furthermore, out of one natural sample, we simultaneously obtain enzymes probably derived from different bacterial strains, growing in the same environment.
This work was supported by New England Biolabs, Inc. 240 County Road, Ipswitch, MA 01938, USA.
The authors have not declared any conflict of interests.