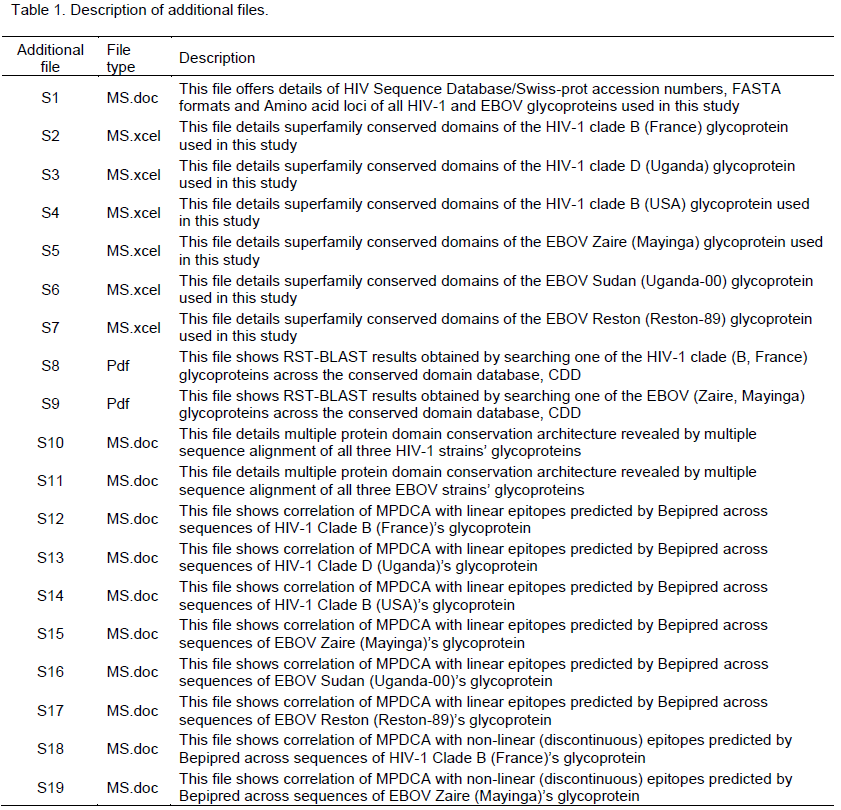
ABSTRACT
Epitope prediction is a critical step to diagnostic and vaccine discovery. Despite existence of some parameters for epitope discovery, this area remains inconclusive and wanting-for new complementary or stand-alone tools. The phenomenon of multiple protein-domain conservation architecture (MPDCA) as used here refers to homologous motifs unveiled by multiple sequence alignments across strain-variants of the same protein aside of the conserved domains (CD) present within the same super family. Unpublished data suggests that MPDCA might be a confounder of epitope necessitating further investigation as a predictor of the same. The ease of determining MPDCA is appealing when considering protein-analysis; specifically epitope discovery. This study aimed to validate MPDCA as a predictive confounder of epitope. Using two-sets of surface viral glycoproteins of human immunodeficiency virus type I, HIV-1 (gp120) and Ebola virus, EBOV (gp1,2 preprotein) (selected because their CD-architecture has widely been studied, their sequences are available in public databases, and the same are well annotated), the MPDCAs among three different virus-strains in each-set, were compared to epitopes predicted by established tools (Bipred and DiscoTope). 4/6 (66.6%) of the linear epitopes confounded MPDCA, with 3/6 (50%) of these MPDCA’s confounding with the predicted linear epitopes (LE) at identities of > 50%, when compared to just 3/6 (50%) of the discontinuous epitopes (DE) that confounded with MPDCA at a < 50% identity. MPDCA is a non-deterministic confounder of Linear B cell epitopy. There is no causal relationship between the two, much as there is an evident co-occurrence. Therefore, MPDCA cannot accurately be used as an additional parameter to predict linear and or non-linear B cell epitopes.
Key words: Epitope, multiple protein-domain conservation architecture (MPDCA), discontinuous epitopes (DE).
Protein-epitopes or antigenic determinants are surface situated protein-motifs that are recognized by either the B or T cell arm of the immune system. Protein-epitopes can either be conformational (non-linear, discontinuous) or linear(Huang and Honda, 2006). Identifying epitopes of particular pathogen-proteins, represents a critical step in the discovery of diagnostics and vaccines for infectious diseases. As a consquence, several groups have previously focused on uncovering the biophysical determinants of epitope (Korber et al., 2006). Despite the rigorous inquest to which the subject of epitope prediction has been subjected, the accurate prediction of epitope remains incomplete (Korber et al., 2006; Emini et al., 1985; Chou and Fasman, 1978; Haste Andersen et al., 2006; Karplus and Schulz, 1985; Kolaskar and Tongaonkar, 1990; Larsen et al., 2006; Parker et al. 1986; Zhang et al., 2008). New parameters are sought to complement or even replace the existing ones as a strategy to enhance the process of epitope prediction. Proteins belonging to a particular super family are defined by the presence of conserved domains (CD) therein. CD have previously been grouped together into a conserved domain database (CDD) as a strategy to allow easy annotation of newly sequenced proteins (Sievers et al., 2011; Geer et al., 2002; Marchler-Bauer et al., 2011). On the contrary, multiple sequence alignments of variants of the same protein from say different pathogen-strains within the same species (which are thereby homologous) reveals the occurence of 100% identical sequence-conservation which is not necessarily of CD nature (Sievers et al., 2011). Henceforth, we chose to refer to this phenomenon as ‘multiple protein-domain conser-vation architecture (MPDCA)’. We have co-incidentally previously uncovered a repetitive occurence of B cell epitope within the context of MPDCA (Unpublished data), findings which have prompted us to question if MPDCA may be a confounder useful towards epitope prediction. Such quest is justified by the fact that MPDCA is an easy and fast parameter to investigate which if proven to be predictive of epitopy, will simplify vaccine or diagnostic discovery.
This study aimed to validate MPDCA as a predictive confounder of epitopy. To do so, we used two-sets of surface viral glycoproteins of human immunodeficiency virus type I, HIV-1 (gp120) and Ebola virus, EBOV (gp1,2 preprotein) (selected because their CD-architecture has widely been studied, their sequences are available in public databases, and the same are well annotated). The MPDCAs in these two-sets of viral glycoproteins among three different virus-strains in each-set, were compared to epitopes predicted by established tools (Bipred and DiscoTope). The authors report results to confirm a non-deterministic confounding of MPDCA with linear B cell epitope (LC); but no definitive correlation with discontinuous epitope (DE).
This study was limited to in-silico sequence analyses, which did not necessitate the author to seek ethical approval from his institutional review board(s). All analyses presented below where done using default settings of software and databases described.
Identifying MPDCAs of the case-study HIV-1 and EBOV strain glycoproteins
Affirming super-family evolutionary ancestory across the case-study viral glycoproteins
Design: In-silico sequence analysis.
Software, databases and sequences: Conserved domain architecture retrieval tool (cDART) (Geer et al., 2002), reverse position-specific (RPS)- basic local sequence alignment tool (BLAST) (Altschul et al. 1997), the conserved domain database (Marchler-Bauer et al., 2011), accession # of the case-study viral glycoproteins of HIV [HIV-1 Clade B (France) SP “|Q00A19|” corresponding to HIVSeqDB “|DQ863898|” (Labrosse et al., 2006); HIV-1 Clade D (Uganda) SP “|D0EMT9|” corresponding to HIVSeqDB “Z19524” (Bruce et al., 1993); HIV-1 Clade B (USA) SP “|D0EMT9|” corresponding to HIVSeqDB “|GQ859302|” (Liu et al., 2009); and EBOV [EBOV Zaire (Mayinga) SP “|Q05320|” (Sanchez et al., 1993); EBOV Sudan (Uganda-00) SP“|Q7T9D9|” (Sanchez et al., 2004); EBOV Reston (Reston-89) SP “|Q66799|” (Sanchez et al., 1996).
The details of amino acid sequences are listed in supporting file S1.
Intervention: We searched the CDD for conserved domains (CD) by feeding the accession # of the respective case-study viral glycoproteins into the RPS-BLAST linked to the CDD as per user guide.
Measured variables: CD specific to the viral glycoprotein super-family.
Unveiling MPDCAs among the case-study viral glycoproteins
Design: In-silico multiple sequence analysis
Software, databases and sequences: Clustal Omega (Sievers et al., 2011), and FASTA format amino acids (Aa) sequences of the case-study viral glycoproteins of HIV-1 and EBOV were used are details are shown in supporting file S1 (Table 1).
Intervention: Multiple sequence alignments of the above HIV-1 and EBOV glycoprotein was done by individually feeding the FASTA formats of the individual virus-group’s (either HIV-1 or EBOV) glycoproteins’ amino acid sequences into the Clustal omega software at default setting.
Measured variables: MPDCA were defined as peptides of > 6 Aa length and cross-strain homology of 100%. Peptides of > 6 Aa were selected, as this is the recognized minimum peptide length with variable reconstructable immunogenicity in-vivo (Emini et al., 1985; Chou and Fasman, 1978; Haste Andersen et al., 2006; Karplus and Schulz, 1985; Kolaskar and Tongaonkar, 1990; Larsen et al., 2006; Parker et al. 1986; Zhang et al., 2008).
Exposing B cell epitopes within the case-study viral glycoproteins
Linear B cell epitope prediction by bipred
Design: Immuno-informatics.
Software, databases and sequences: Bepipred linear B cell prediction software (Larsen et al., 2006) and FASTA format amino acids (Aa) sequences of the case-study viral glycoproteins of HIV-1 and EBOV are detailed in supporting file S1 (Table 1).
Intervention: Linear B cell epitopes were derived by feeding the FASTA formats of the amino acids (Aa) sequences of the case-study viral glycoproteins of HIV-1 and EBOV into the Bepipred user interface at default.
Measured variables: Linear B cell epitopes.
Design: Immuno-informatics.
Software, databases and sequences: DiscoTope conformational B cell prediction software (Haste Andersen et al., 2006) and 3-D crystal structure accession # for the case-study viral glycoproteins of HIV-1 and EBOV (PDB entry “3dnl” and “3CSY” respectively).
Intervention: Conformational B cell epitopes were derived by individually feeding the PDB entry accessions of the case-study viral glycoproteins of HIV-1 and EBOV into the DiscoTope user interface at default.
Measured variables: Conformational B cell epitopes.
Correlating multiple domain conservation architectures with predicted epitope among the case-study vira; glycoproteins
This was more of a mathematical or statistical analysis of the data above; with a focus on ascertaining the correlation between epitopes and MPDCA. The few cases used for this validation stage could not permit derivation of variation coefficients with statistical significance. Instead, a cross-tabulation of MPDCA with either linear epitope (LE) or discontinuous epitope (DE) was done.
Availability of supporting data
"The data set(s) supporting the results of this article is (are) included within the article (and its additional file(s)" (Table 1).
The results confirm a non-deterministic confounding of MPDCA with linear B cell epitope (LC); and no definitive correlation with discontinuous epitope (DE).
MPDCA among EBOV and HIV-1 strain glycoproteins
First, the presence of conserved-domains consistent with the viral glycoprotein super-families studied was affirmed and subsequently unveiled multiple protein domain conservation architectures across the same case-study viral glycoproteins.
Super-family conserved domains (CD) across the case-study viral glycoproteins
All case-study viral glycoproteins were affirmed to lie within the HIV-1 and EBOV super-family of glycoproteins (for details, see additional files S1, S2, S3, S4, S5, S6, and S7) (Table 1). A phylogenetic illustration of this evolutionary ancestry across the HIV-1 case study glycoproteins, gp120 is shown in Figure 1. Schematics of the conserved domain architecture within the case-study viral glycoproteins are shown in the RST-BLAST results detailed in additional supporting files S8 and S9 (Table 1).

These data served to justify our choice of strain-type variants of the study viral glycoproteins within the same species of virus. CD may be viewed as functional protein motifs, which by virtue of their inter-network molecules within a network assume evolutionary patterns of hub-proteins. Therefore as pathogens evolve (presumably across strains in the same species), the CD are maintained to sustain their functionality. Further, because CDs are functional motifs which must interact with ligands in the network, the same are often located on the surface, thereby explaining the confounding between CD and epitope (Geer et al., 2002; Marchler-Bauer et al., 2011). The multiple protein-domain conservation architecture (MPDCA) revealed that, on the other hand, may or may not be functional motifs as is elucidated by prior studies and further evidence provided. Nonetheless, it appears that the same MPDCA are under pressure from other interaction within the network; be they functional or structural (proxy).
MPDCA among the case-study viral glycoproteins
Six (6) MPDAs with more than six amino acids (Aa) length and 100% identity were unveiled in each of the case-study glycoprotein groups, HIV-1 gp120 and EBOV gp1,2. The respective details of these findings are shown by different color shades in supporting files S10 and S11 (Table 1). Fraser et al. (2002) previously found that the connectivity of well-conserved proteins in the network is negatively correlated with their rate of evolution. Overall, this group showed that proteins with more interactions evolve more slowly not because they are more important to the organism, but because a greater proportion of the protein is directly involved in its function. In contrast to this claim that proteins with more interaction partners (sometimes called hubs) are- owing to an assumed high density of binding sites, both physiologically more important and slow evolving; Batada et al. (2006) found that hub proteins are indeed more important for cellular growth rate and are under tight regulation but are not slow evolving. These studies suggest that at sites important for interaction between proteins (such as the MPDCA which were studied), evolutionary changes occur largely by coevolution, in which substitutions in one protein result in selection pressure for reciprocal changes in interacting partners. He argued that, in the same manner as evolutionary changes in the B cell paratrope could potentially influence epitope architecture, MPDCA may be under pressure from their indeterminate interactions. Such analogy to hub proteins with regards to the conservation patterns of protein domains is, however, debatable. Specifically, the primary reason for conservation of sequence across a domain is preservation of the domain fold and secondary structure elements (internal interactions). Conservation of active site residues and structure, and conservation of binding regions contribute to local sequence conservation (that is, within the same functional sub-family). Also, the assumption of a co-evolutionary model may not be universal, since this will only occur when the domain interacts with another protein (or DNA/RNA segment). However, most domain function (enzymes, signalling) involve interactions with small molecules which do not "evolve". Also, protein domains occur in proteins throughout the cell, and are not predominantly associated with cell walls or membranes (Huang and Honda, 2006; Korber et al., 2006; Emini et al., 1985; Chou and Fasman, 1978; Haste Andersen et al., 2006; Karplus and Schulz, 1985; Kolaskar and Tongaonkar, 1990; Larsen et al., 2006; Parker et al. 1986; Zhang et al., 2008; Sievers et al., 2011; Geer et al., 2002; Marchler-Bauer et al., 2011; Fraser et al., 2002; Batada et al., 2006; Altschul et al., 1997; Labrosse et al., 2006; Bruce et al., 1993; Liu et al., 2009; Sanchez et al., 1993; Sanchez et al., 2004; Sanchez et al., 1996).
B cell epitopes within the case-study viral glycoproteins
We uncovered both linear and non-linear B cell epitopes in all case-study viral glycoproteins as is further detailed below.
Linear B cell epitopes prediction by bepipred
Several linear B cell epitopes were unveiled in all case-study viral glycoproteins that are shown further in supporting files S12, S13, S14, S15, S16 and S17 (Table 1).
Non-linear B cell epitopes prediction by discotope
The conformational B cell epitopes unveiled across the case- study viral glycoproteins are shown further in supporting files S18 and S19 (Table 1), respectively for HIV-1 and EBOV.
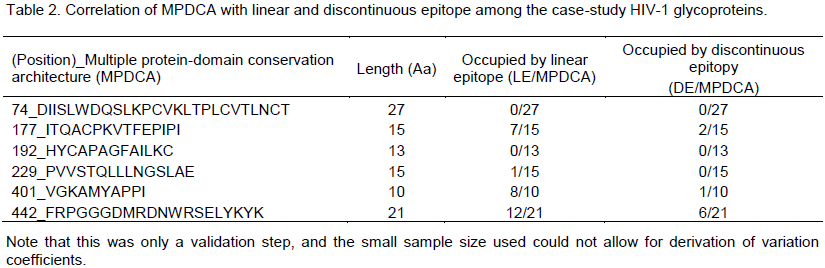
Correlation of MPDCA and predicted epitope among the case-study viral glycoproteins
The author observed an arbitrarily non-deterministic confounding of MPDCA with linear B cell epitope (LC); but no definitive correlation with discontinuous epitope (DE). Specifically, 4/6 (66.6%) of the linear epitopes confounded MPDCA, with 3/6 (50%) of these MPDCA’s confounding with the predicted linear epitopes (LE) at identities of > 50% (Tables 1 and 2) when compared to just 3/6 (50%) of the discontinuous epitopes (DE) that confounded with MPDCA at a < 50% identity (Tables 2 and 3). There are several weaknesses in our approach and findings. Since this was only a first-step in proof of concept, and the small sample size used (there are over 100,000 sequences for HIV GP120) could not allow for derivation of variation coefficients, he argued that further expanded work is sought in this direction to better inform the performance of MPDCA. Further, the methods to which he compare the performance of MPDCA as a predictor of B cell epitope, have variable precision and are not necessary the best (Emini et al., 1985; Chou and Fasman, 1978; Haste Andersen et al., 2006; Karplus and Schulz, 1985; Kolaskar and Tongaonkar, 1990; Larsen et al., 2006; Parker et al. 1986; Zhang et al., 2008). Certainly in the case of the linear predictor the false positive prediction rate is very high making it unusable as a benchmark (no false negative rate given, because not much could be determined as this stage).
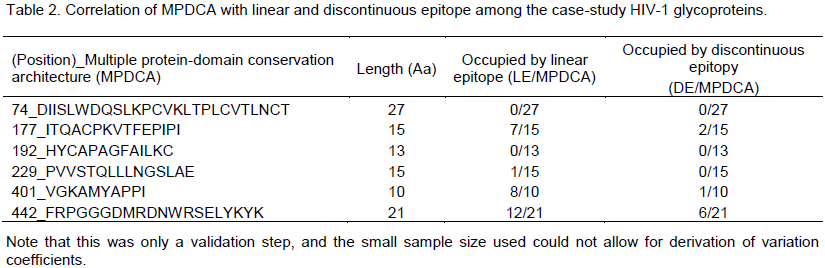
Overall, these data show that MPDCA is a non-deterministic confounder of linear B cell epitope. Moreover, there appears to be no causal relationship between the two, much as there is an evident co-occurrence. Therefore, MPDCA cannot accurately be used as an additional parameter to predict linear and or non-linear B cell epitopes. The only possible applicability of MPDCA in epitope discovery is that of rapidly scanning across proteins to see areas that may be potentially epitopic. More important to note outside of our findings is that MPDCA cannot predict antigenicity or immunogenicity. Further, another major shortcoming with using MPDCA to predict linear epitopy, is that MPDCA have the potential to interact with other players in the network, a behavior that might mask or even conceal their architecture in-vivo, making them inappropriate vaccine or diagnostic targets.
The author declare that there is no conflict of interest.
The author would like to thank Prof. Wilson Byarugaba, Prof. Moses L. Joloba, Prof. Joseph Olobo, Prof. Deogratius Kaddu-Mulindwa, Dr. Henry Kajumbula (all at Makerere University College of Health Sciences, Kampala, UG), Prof. Leslie Lobel, Dr. Julius J. Lutwama and Dr. Robert Downing (CDC/UVRI, Kampala, UG) for a keen interest on prior studies in this direction. This and related work was supported by the Government of Canada through Grand Challenges Canada “ Round IV Rising Stars in Global Health [Grant # S4_0280-01] to M.W. “ Transition-To-Scale” [Grant # TTS-0709-05].
MPDCA, Multiple protein-domain conservation architecture; CD, conserved domains; DE, discontinuous epitopes; LE, linear epitopes; CDD, conserved domain database; cDART, conserved domain architecture retrieval tool; RPS, reverse position-specific; BLAST, basic local sequence alignment tool.
REFERENCES
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997).Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402.
Crossref |
|
|
Batada NN, Hurst LD, Tyers M (2006). Evolutionary and physiological importance of hub proteins. Plos Comput. Biol. 2(7): e88.
Crossref |
|
|
Bruce C, Clegg C, Featherstone A, Smith J, Oram J (1993). Sequence analysis of the gp120 region of the env gene of Ugandan human immunodeficiency proviruses from a single individual. AIDS Res. Hum. Retrovir. 9 (4):357-363.
Crossref |
|
|
Chou PY, Fasman GD (1978) Prediction of the secondary structure of proteins from their amino acid sequence. Adv. Enzymol. Relat. Areas Mol. Biol. 47:45-148.
PMid:364941 |
|
|
Emini EA, Hughes JV, Perlow DS, Boger J (1985). Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 55(3):836-839.
Pubmed |
|
|
Fraser HB, Hirsh AE, Steinmetz LM, Scharfe C, Feldman MW (2002). Evolutionary Rate in the Protein Interaction Network. Science 296:750-752
Crossref |
|
|
Geer LY, Domrachev M, Lipman DJ, Bryant SH (2002). CDART: protein homology by domain architecture. Genome Res. 12(10):1619-623.
Crossref |
|
|
Haste Andersen P, Nielsen M, Lund O (2006). Prediction of B-cell epitopes using protein 3D structures. Protein Sci. 15(11): 2558-2567.
Crossref |
|
|
Huang J, Honda W (2006). CED: a conformational epitope database. BMC Immunol. 7: 7
Crossref |
|
|
Karplus PA, Schulz GE (1985). Prediction of Chain Flexibility in Proteins - A tool for the Selection of Peptide Antigens. Naturwissenschafren. 72: 212-213.
Crossref |
|
|
Kolaskar AS, Tongaonkar PC (1990). A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 276(1-2):172-174.
Crossref |
|
|
Korber B, LaBute M, Yusim K (2006). Immunoinformatics comes of age. PLoS Comput Biol. 2(6): e71.
Crossref |
|
|
Labrosse B, Morand-Joubert L, Goubard A, Rochas S, Labernardière JL, Pacanowski J, Meynard JL, Hance AJ, Clavel F, Mammano F(2006). Role of the envelope genetic context in the development of enfuvirtide resistance in human immunodeficiency virus type 1-infected patients. J. Virol. 80(17):8807-8819.
Crossref |
|
|
Larsen JE, Lund O, Nielsen M (2006). Improved method for predicting linear B-cell epitopes. Immunome Res. 2:2.
Crossref |
|
|
Liu Y, Woodward A, Zhu H, Andrus T, McNevin J, Lee J, Mullins JI, Corey L, McElrath MJ, Zhu T (2009). Preinfection human immunodeficiency virus (HIV)-specific cytotoxic T lymphocytes failed to prevent HIV type 1 infection from strains genetically unrelated to viruses in long-term exposed partners. J. Virol. 83 (20): 10821-10829.
Crossref |
|
|
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH(2011). CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39(Database issue): D225-229.
Crossref |
|
|
Parker JM, Guo D, Hodges RS (1986). New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 25(19): 5425-5432.
Crossref |
|
|
Sanchez A, Kiley MP, Holloway BP, Auperin DD (1993). Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 29: 215-240.
Crossref |
|
|
Sanchez A, Lukwiya M, Bausch D, Mahanty S, Sanchez AJ, Wagoner KD, Rollin PE (2004). Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: cellular responses, virus load, and nitric oxide levels. J. Virol. 78: 10370-10377.
Crossref |
|
|
Sanchez A, Trappier SG, Mahy BW, Peters CJ, Nichol ST (1996). The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U.S.A. 93:3602-3607.
Crossref |
|
|
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 7:539.
Crossref |
|
|
|
Zhang Q, Wang P, Kim Y, Haste-Andersen P, Beaver J, Bourne PE, Bui HH, Buus S, Frankild S, Greenbaum J, Lund O, Lundegaard C, Nielsen M, Ponomarenko J, Sette A, Zhu Z, Peters B (2008). Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 36(Web Server issue):W513-518. |